Likelihood and Substitution Models
1. Problems with Parsimony
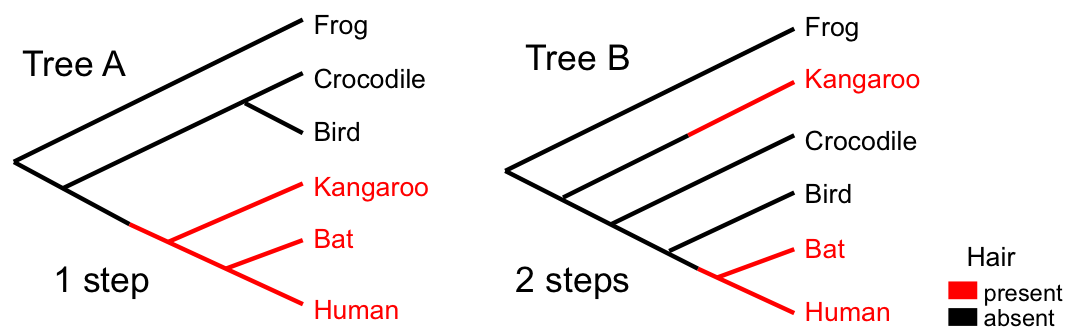
While parsimony is intuitive and computationally tractable for small problems, it has several limitations:
- "Large parsimony problem" is computationally expensive
- NP-hard problem requiring heuristic search algorithms
- No guarantee of finding global optimum
- Gives point estimates with no uncertainty quantification
- No confidence intervals or support values
- No way to compare alternative hypotheses statistically
- Questionable biological basis
- Assumes all changes are equally unlikely
- Ignores branch lengths
- Not model-based
- No formal hypothesis testing
- No model comparison
- Hidden problems
- Long branch attraction
- Inconsistency under certain conditions
2. Modelling Neutral Sequence Evolution
Why Use Models?
We need a model to relate what we observe (data) to what we want to know (hypotheses and parameters).
We use probabilistic models for molecular evolution because:
- We don't know enough about mutation mechanisms for deterministic models
- Stochastic effects are important at molecular level
- Allows uncertainty quantification
- Enables formal statistical inference
Genetic Distance: From p-distance to Model-based Estimates
The p-distance
The proportion of sites that differ between two aligned sequences.
Example: If 15 out of 100 sites differ, p-distance = 0.15
- Simple to calculate
- Always between 0 and 1
- Also called normalized Hamming distance
- BUT: underestimates true evolutionary distance (we'll see why)

p-distance Example

p-distance = 3/7 ≈ 0.43
Interpretation: 43% of sites differ between these sequences
Why p-distance Underestimates True Distance
The p-distance usually underestimates the true genetic (evolutionary) distance because it doesn't account for:

Biological Intuition
Imagine watching a bird feeder at different times:
- You see a korimako/bellbird at 9am and you again see a korimako/bellbird at 5pm.
- Same bird all day? Or did multiple birds use the spot?
- Without continuous observation, you can't tell!
Similarly, DNA sites may have changed multiple times between ancestors and descendants.
- Multiple substitutions: Same site changing more than once
- Parallel substitutions: Same change occurring independently
- Back-substitutions: Reversions to ancestral state
Relationship Between p-distance and Genetic Distance
Why does p-distance saturate at 0.75?
With 4 nucleotides and random substitutions:
- Probability of staying the same = 1/4
- Probability of being different = 3/4
- After infinite time, sequences become random with respect to each other
- Random sequences differ at 75% of sites
3. Continuous-Time Markov Chains (CTMCs)
Mathematical Framework
A stochastic process where:
- State X(t) is a function of continuous time
- Future states depend only on current state (Markov property)
- Transition rates are constant over time
What does this mean biologically?
A continuous-time Markov chain models how nucleotides change over time:
- Each site evolves independently
- Changes can happen at any time
- The probability of change depends only on current state, not history
- Like radioactive decay - constant probability of change per unit time
Example: An 'A' nucleotide doesn't "remember" how long it's been an 'A' - it always has the same probability of mutating in the next instant.
CTMCs obey the Chapman-Kolmogorov equation:
In plain English: To get from state X(t₀) to X(t₁), sum over all possible paths through intermediate states.
Mathematical Details (Optional)
The differential form (Master equation/Kolmogorov forward equation):
Where Q is the instantaneous rate matrix with:
- \(Q_{ij}\) = rate of change from state i to j (i ≠ j)
- \(Q_{ii} = -\sum_{j \neq i} Q_{ij}\) (rows sum to zero)
Relationship Between Q and P(t)
The transition probability matrix P(t) is the matrix exponential of Qt:
Intuition for the Matrix Exponential
This formula tells us:
- P(0) = I (no time = no change)
- P(small t) ≈ I + tQ (short time = approximately linear)
- P(large t) → stationary distribution (long time = equilibrium)
Think of it as compound interest for mutations!
CTMC Example
Two-State System
Consider a simplified DNA with only purines (R) and pyrimidines (Y):
This means:
- Rate R→Y: 2 per unit time
- Rate Y→R: 1 per unit time
Biological interpretation: Transitions (R↔Y) happen at different rates in each direction
Time spent in state before transition follows exponential distribution:
Time-Reversibility and Detailed Balance
A CTMC is time-reversible if it satisfies detailed balance:
where π is the stationary distribution.
For time-reversible CTMCs:
- Process looks the same forward and backward in time
- Stationary distribution satisfies: \(\pi Q = 0\)
- Mathematical convenience for phylogenetics
- Important: No biological justification required!
4. DNA Substitution Models
Now let's see how CTMCs are used to model DNA evolution. We'll start simple and build complexity.
Jukes-Cantor (JC69) Model
The Simplest Model

Assumptions:
- All substitutions equally likely
- Equal base frequencies (25% each)
- One parameter: μ (overall rate)
Rate matrix:
Transition probabilities:
Genetic Distance Under JC69
Given observed p-distance, we can estimate the true genetic distance:
- Formula undefined if p ≥ 3/4 (sequences too divergent)
- Assumes all changes equally likely (unrealistic)
- Assumes equal base composition (often violated)
Kimura 2-Parameter (K80) Model
Adding Biological Realism

Key innovation: Transitions ≠ Transversions
- Transitions (α): Purine↔Purine, Pyrimidine↔Pyrimidine
- Transversions (β): Purine↔Pyrimidine
- Usually α > β (transitions more common)
Rate matrix:
Why are transitions more common?
Chemical similarity:
- Purines (A,G): Two-ring structures
- Pyrimidines (C,T): One-ring structures
- Replacing like-with-like causes less structural disruption
HKY Model
Hasegawa-Kishino-Yano (1985)

Features:
- Different transition/transversion rates
- Unequal base frequencies (πA, πC, πG, πT)
- Reflects GC-content variation
Rate matrix:
General Time Reversible (GTR) Model
The Most General Model

Features:
- All substitution types can have different rates
- 6 rate parameters + 4 frequency parameters
- Most flexible time-reversible model
- Includes all previous models as special cases
Rate matrix:
Model Comparison Summary
| Model | Parameters | Key Feature | When to Use |
|---|---|---|---|
| JC69 | 1 | All equal | Very similar sequences |
| K80 | 2 | Ts/Tv ratio | Moderate divergence |
| HKY | 5 | +Base frequencies | GC-content varies |
| GTR | 9 | All different | Complex datasets |
Rate Heterogeneity Among Sites
Biological Reality
Not all sites in a gene evolve at the same rate:
- Active sites: Highly conserved (slow evolution)
- Structural regions: Moderate constraints
- Surface loops: Fewer constraints (fast evolution)
- Synonymous sites: Often evolve fastest
We model rate variation using a gamma distribution:

Understanding the shape parameter α
- α < 1: Most sites evolve slowly, few evolve very fast
- α = 1: Exponential distribution
- α > 1: Bell-shaped distribution
- α → ∞: All sites evolve at same rate
Model Comparison Example
HIV Distance Estimates
HIV-1B vs other strains (env gene):
| Comparison | p-distance | JC69 | K80 | Tajima-Nei |
|---|---|---|---|---|
| HIV-O | 0.391 | 0.552 | 0.560 | 0.572 |
| SIVcpz | 0.266 | 0.337 | 0.340 | 0.427 |
| HIV-1C | 0.163 | 0.184 | 0.187 | 0.189 |
Observation: Model choice matters more for divergent sequences!
5. Likelihood-Based Phylogenetic Inference
From Distances to Likelihood
Distance methods lose information by summarizing sequences as pairwise distances. Likelihood methods use all the data.
The Likelihood Function
The likelihood for parameter θ under model M given data D is:
In words: "The probability of observing our data if the parameter values were θ"
Important: The likelihood is NOT a probability distribution over θ!
Simple Example: Coin Flips
5 tosses give: D = (H,T,T,H,T)
What's the probability of getting exactly this sequence?
This is the likelihood function L(f|D)!
Maximum Likelihood Inference
The maximum likelihood estimate is the parameter value that makes the observed data most probable.
Why Maximum Likelihood?
- Intuitive: Choose parameters that make data most likely
- Optimal properties: Consistent, efficient (with enough data)
- General framework: Works for any statistical model
- BUT: Only gives point estimates, no uncertainty!
Tree Likelihood
For phylogenetics, we want P(alignment | tree, model):

The likelihood calculation involves:
- For each site in the alignment:
- Consider all possible ancestral states
- Calculate probability of observing tip states
- Sum over all possibilities
- Multiply across sites (assumes independence)
Felsenstein's Pruning Algorithm
Joseph Felsenstein (1973) solved this using dynamic programming:
The Pruning Algorithm
Key idea: Work from tips toward root, storing partial likelihoods
Define: Lk(s) = likelihood of subtree below node k if node k has state s
For leaf nodes:
For internal nodes with children i and j:
Where P(x|s,t) is the probability of state s changing to x in time t
Why This Works
Instead of considering all 4^(n-1) combinations:
- Store 4 numbers at each node (partial likelihoods)
- Build up from tips to root
- Reuse calculations (dynamic programming principle)
Result: Linear time in number of taxa!
Maximum Likelihood Software
ML Phylogenetic Software
- IQ-TREE (Nguyen et al., 2015)
- State-of-the-art tree search
- Automatic model selection
- Ultrafast bootstrap
- RAxML (Stamatakis, 2014)
- Highly optimized for large datasets
- Excellent parallelization
- Standard in many pipelines
- PhyML (Guindon et al., 2010)
- User-friendly
- Good for moderate datasets
- Fast approximate likelihood ratio tests
- FastTree (Price et al., 2009)
- Approximate but very fast
- Can handle millions of sequences
- Good for initial analyses
Limitations of Maximum Likelihood
- No confidence intervals
- How certain are we about the tree?
- Bootstrap provides some help (but it's ad hoc)
- Model selection challenges
- How do we choose between JC69, K80, HKY, GTR?
- Likelihood ratio tests only work for nested models
- Can't incorporate prior knowledge
- What if we know some clades are unlikely?
- What if we have fossil calibrations?
We need a framework that handles uncertainty properly → Bayesian inference!
Summary
This lecture introduced model-based phylogenetic inference:
- Why models?
- Parsimony has serious limitations
- Models enable statistical inference
- Can test hypotheses formally
- Substitution models as CTMCs:
- Continuous-time Markov chains model sequence evolution
- Models range from simple (JC69) to complex (GTR+Γ)
- More complex models fit data better but risk overfitting
- Key biological insights:
- p-distance underestimates true distance due to multiple hits
- Transitions more common than transversions
- Base composition varies across genomes
- Different sites evolve at different rates
- Likelihood methods:
- Use all information in the alignment
- Felsenstein's algorithm makes computation feasible
- BUT: still only point estimates
- Next step: Bayesian inference provides a complete framework for uncertainty
Key Takeaways
- For biologists: Models let us correct for unseen changes and test evolutionary hypotheses
- For computer scientists: Phylogenetics offers rich algorithmic challenges (DP, matrix exponentials, tree search)
- For everyone: Understanding models is crucial for interpreting phylogenetic results
- Felsenstein (2004) "Inferring Phylogenies" - Chapters 13-16
- Yang (2014) "Molecular Evolution: A Statistical Approach" - Chapters 1-2
- Drummond & Bouckaert (2015) "Bayesian Evolutionary Analysis with BEAST" - Chapter 2
Check Your Understanding
- Why does p-distance underestimate true evolutionary distance?
- What biological reasons explain why transitions are more common than transversions?
- How does Felsenstein's algorithm achieve computational efficiency?
- What are the key differences between JC69, K80, HKY, and GTR models?
- Why isn't likelihood a probability distribution over parameters?