Maximum Parsimony
1. Characters and Character States
To understand parsimony, we first need to understand the fundamental units of phylogenetic analysis: characters and character states.
- Character: A feature that can vary among organisms (e.g., a nucleotide position in DNA, presence/absence of wings)
- Character state: The specific condition of a character in a particular organism (e.g., A, C, G, or T at a DNA position)
- Character matrix: A table showing character states for all taxa across all characters
Example character matrix for DNA sequences:
| Taxon | Character (position) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| Human | A | G | C | T | A | C | G | G | T | A |
| Chimp | A | G | C | T | A | C | G | A | T | A |
| Gorilla | A | G | T | T | G | C | G | A | T | C |
| Orangutan | A | A | T | C | G | T | A | A | C | C |
Similarities and differences in character states provide evidence for inferring evolutionary relationships.
When Do Characters Support the Correct Tree?
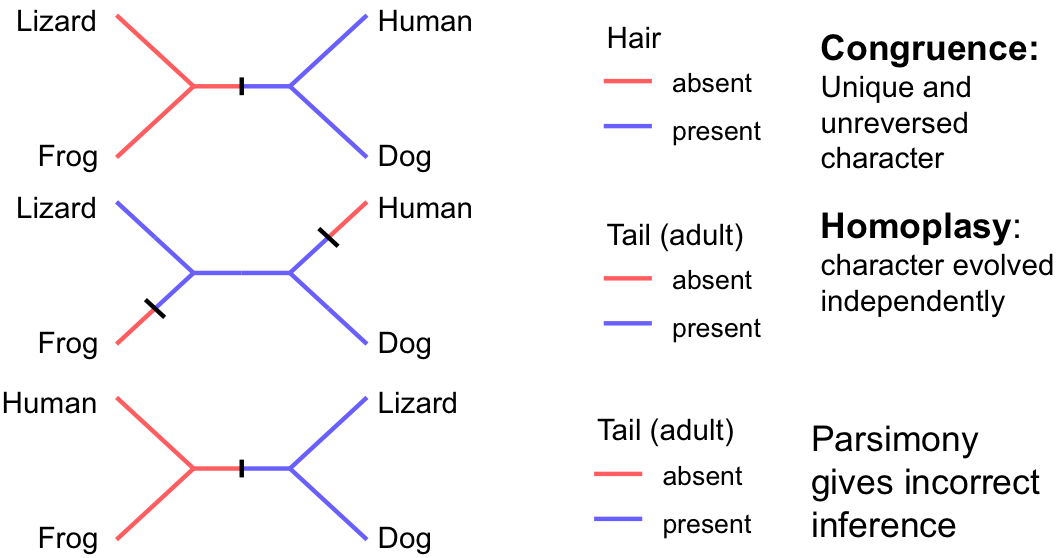
Characters support correct phylogenetic inference when:
- Shared character states reflect common ancestry (homology)
- Changes are rare enough that multiple changes to the same state are uncommon
- There's sufficient variation to be informative
The Problem of Homoplasy
Similarity in character states that is not due to common ancestry. This can arise from:
- Convergent evolution: Independent evolution of similar traits
- Parallel evolution: Independent evolution along similar pathways
- Reversals: Return to an ancestral character state
Reversals
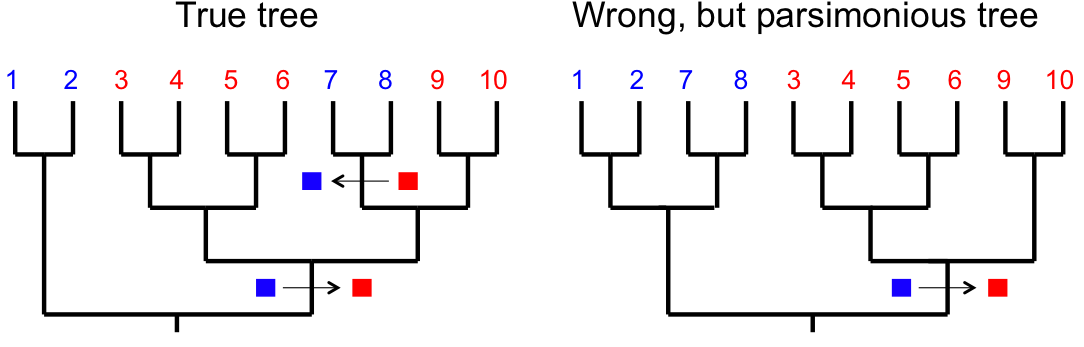
If a character reverts to an ancestral state, this can affect phylogenetic inference by creating false signals of relationship.
2. The Parsimony Principle
A method of phylogenetic inference that selects the tree(s) requiring the fewest evolutionary changes to explain the observed data.
Key concepts in parsimony analysis:
- Key issue: How to separate homoplasy from homology (true shared ancestry)
- Parsimony criterion: Favors hypotheses that maximize congruence and minimize homoplasy
- Optimization problem: Find the tree with the minimum number of character state changes
Character Fit to Trees
The "fit" of a character to a tree is defined as the minimum number of steps (changes) required to explain the observed distribution of character states at the tips.
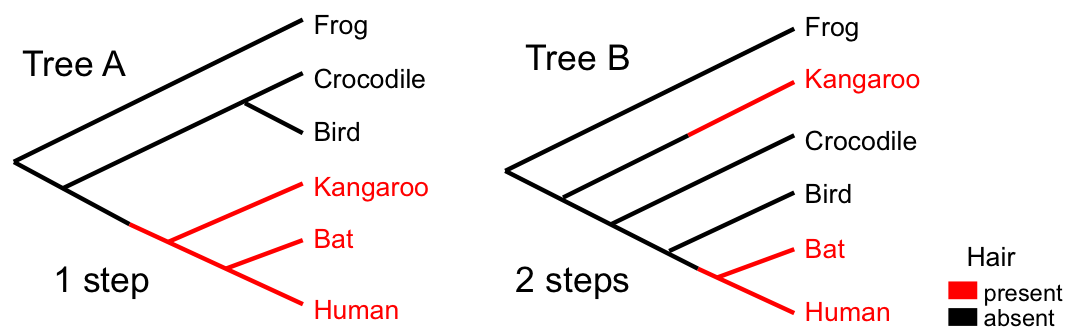
Parsimony Calculation
Given a set of characters (e.g., aligned sequences):
- For each character, determine the minimum number of steps on a given tree
- Sum over all characters to get the tree length
- The most parsimonious trees (MPTs) have the minimum tree length
Parsimony Informative Sites
Not all characters contribute equally to distinguishing between alternative trees:
- At least two different character states
- Each state appearing in at least two taxa
Examples of non-informative sites:
- Invariant sites: All taxa have the same state (always score 0)
- Singleton sites: Only one taxon differs (always score 1)
Identifying Informative Sites
| Site | Pattern | Informative? | Reason |
|---|---|---|---|
| 1 | AAAA | No | Invariant |
| 2 | AAAG | No | Singleton |
| 3 | AAGG | Yes | Two states, each in ≥2 taxa |
| 4 | AGTC | No | No state in ≥2 taxa |
3. Computing Parsimony Scores
The Small Parsimony Problem
Given a tree topology and character data at the tips, find:
- The minimum number of changes required for each character
- The ancestral states that achieve this minimum
Dynamic Programming Solution
We can solve this efficiently using dynamic programming (Sankoff algorithm):
Sankoff Algorithm
For each node $v$ and each possible state $X$, calculate $m[v,X]$ = minimum cost of the subtree rooted at $v$ if $v$ has state $X$.
For leaf nodes:
For internal nodes (with children L and R):
where $c(X,Y)$ is the cost of changing from state $X$ to state $Y$.

Complexity Analysis
For the small parsimony problem:
- Time complexity: $O(nS^2L)$
- $n$ = number of taxa (gives $2n-1$ nodes in rooted binary tree)
- $S$ = number of possible states (4 for DNA)
- $L$ = sequence length
- At each node: $O(S^2)$ calculations
- Space complexity: $O(nS)$ to store the dynamic programming table
Fitch Parsimony
For the special case where all changes have equal cost (unweighted parsimony), Fitch (1971) developed a faster algorithm:
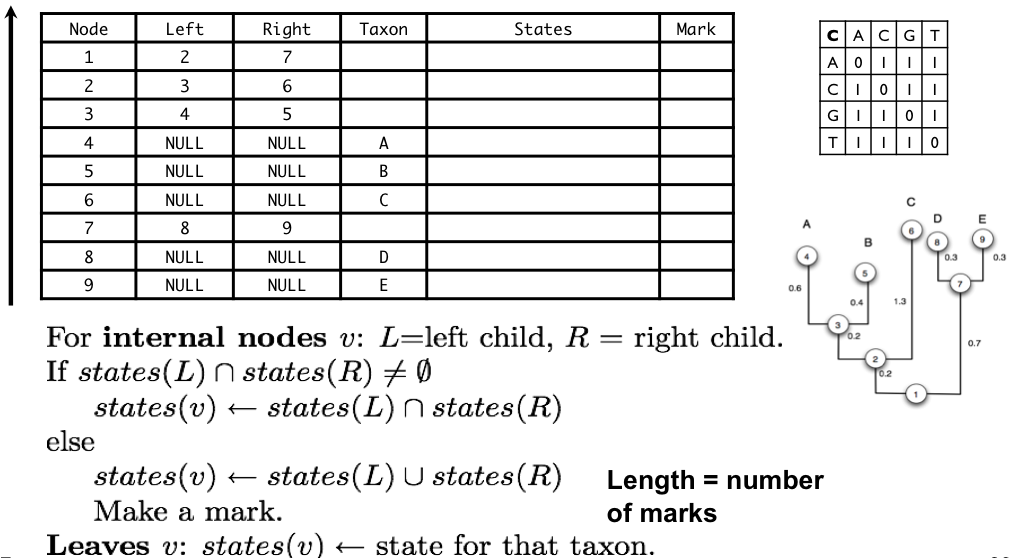
Fitch Algorithm
Phase 1 (Bottom-up):
- For each leaf, assign its observed state
- For each internal node with children having state sets $S_L$ and $S_R$:
- If $S_L \cap S_R \neq \emptyset$: assign $S_L \cap S_R$ (intersection)
- If $S_L \cap S_R = \emptyset$: assign $S_L \cup S_R$ (union) and count one change
Phase 2 (Top-down):
- Assign specific ancestral states using the sets computed in Phase 1
Fitch Algorithm Example
Consider a simple tree with tips having states: ((A,G),(A,T))
- Left child of root: A ∩ G = ∅, so assign {A,G}, cost = 1
- Right child of root: A ∩ T = ∅, so assign {A,T}, cost = 1
- Root: {A,G} ∩ {A,T} = {A}, so assign {A}, no additional cost
- Total parsimony score = 2
4. Finding Optimal Trees
The "large parsimony problem" involves finding the tree topology (or topologies) with the minimum parsimony score:
Search Strategies
- Exhaustive search: Evaluate all possible trees
- Guarantees finding all MPTs
- Only feasible for small numbers of taxa (≤10-12)
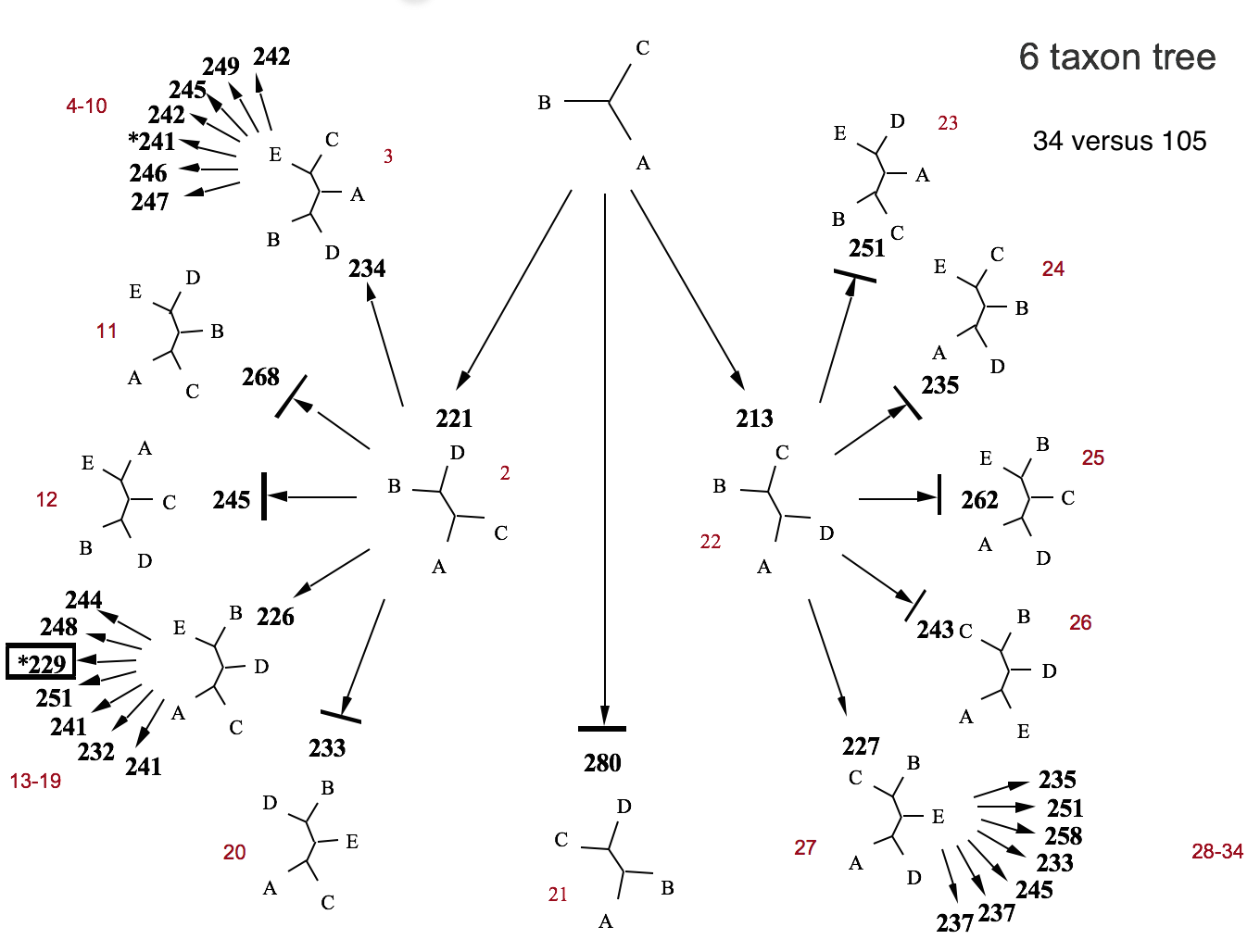
- Branch-and-bound: Intelligent exhaustive search
- Uses lower bounds to eliminate parts of tree space
- Still guarantees finding all MPTs
- Practical for up to ~20-25 taxa
- Heuristic search: Not guaranteed to find optimal trees
- Necessary for larger datasets
- Various strategies for exploring tree space
Branch-and-Bound Example
Heuristic Search Methods
Since the number of trees grows exponentially (NP-complete problem), heuristic methods are essential for larger datasets:
General Heuristic Strategy
- Generate starting tree(s)
- Stepwise addition
- Star decomposition
- Random trees
- Local search via branch swapping
- Nearest Neighbor Interchange (NNI)
- Subtree Pruning and Regrafting (SPR)
- Tree Bisection and Reconnection (TBR)
Branch Swapping Methods
- NNI (Nearest Neighbor Interchange):
- Swaps adjacent branches
- Fastest but most limited
- Can get stuck in local optima
- SPR (Subtree Pruning and Regrafting):
- Detaches a subtree and reattaches elsewhere
- More extensive search than NNI
- TBR (Tree Bisection and Reconnection):
- Breaks tree into two subtrees and reconnects
- Most extensive rearrangements
- Best for escaping local optima
5. Tree Representation in Computers
Understanding how trees are stored in memory is crucial for implementing phylogenetic algorithms:
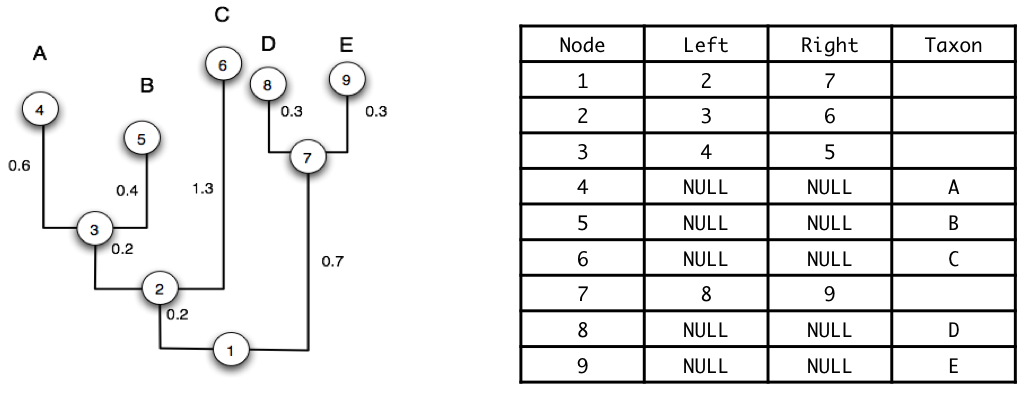
- Pre-order: Visit parent before children (shown in table)
- Post-order: Visit children before parent (used in Fitch algorithm)
Tree Traversal Example
For tree (((A,B),C),(D,E)) with nodes numbered as in the figure:
- Pre-order: 1 → 2 → 3 → 4(A) → 5(B) → 6(C) → 7 → 8(D) → 9(E)
- Post-order: 4(A) → 5(B) → 3 → 6(C) → 2 → 8(D) → 9(E) → 7 → 1
Note: The table in the figure shows nodes in pre-order, which is why they're numbered 1-9 in that sequence.
6. Parsimony Summary
Advantages of Parsimony
- Simple, intuitive principle (Occam's Razor)
- No explicit evolutionary model required
- Fast algorithms for scoring trees
- Can handle any type of character data
- Identifies synapomorphies (shared derived characters)
Limitations of Parsimony
- Can be inconsistent under certain conditions (long branch attraction)
- Doesn't account for multiple substitutions on a branch
- All changes treated equally (unless weighted)
- No measure of uncertainty or support
- No branch length estimation
Key Points to Remember
- The small parsimony problem (scoring a tree) is efficiently solved by dynamic programming
- The large parsimony problem (finding optimal trees) has no efficient solution
- Maximum parsimony finds trees requiring the fewest evolutionary changes
- Not based on an explicit evolutionary model
- Best suited for datasets with low homoplasy
Software for Parsimony Analysis
- PAUP*: Comprehensive phylogenetic analysis with excellent parsimony implementation
- TNT: Optimized for large datasets, very fast
- MEGA: User-friendly interface, good for teaching
- MPBoot: Ultrafast bootstrap approximation for parsimony
Check Your Understanding
- What makes a site parsimony-informative?
- How does the Fitch algorithm differ from the general Sankoff algorithm?
- Why is branch-and-bound better than exhaustive search?
- What is the difference between the small and large parsimony problems?
- Under what conditions might parsimony give misleading results?