Introduction to Phylogenetics
1. Molecules as Documents of Evolutionary History
Molecular sequences contain a wealth of information about evolutionary processes and history. By comparing sequences from different organisms, we can infer their evolutionary relationships.
Example: HIV-1 Sequence Variation
HIV-1 (UK) ATCGGATGCTAAAGCATATGACACAGAGGTACATAATGTTT HIV-1 (USA) ATCAGATGCTAGAGCTTATGATACAGAGGTACA---TGTTT
The differences (highlighted) between these sequences tell us about their evolutionary divergence.
Key points about molecular evolution:
- Macromolecules contain information about the processes and history that formed them
- This information is incomplete, so the full history must be inferred
- Computational biology aims to decipher the information held in molecular sequences about evolutionary processes and history
What is Phylogenetics?
The study of evolutionary relationships among organisms or genes, typically represented as trees showing patterns of descent from common ancestors.
Core concepts in phylogenetics:
- Homology: Similarity due to inheritance from a common ancestor
- Phylogenetic trees: Branching structure representing evolutionary relationships based on common ancestry
- Monophyletic groups (clades): Groups containing species more closely related to each other than to any species outside the group
- Statistical inference: Modern phylogenetics uses probabilistic models of evolution
A Typical Phylogenetic Analysis
- Collect homologous sequences - Identify sequences that share common ancestry
- Construct multiple sequence alignment - Align sequences to identify homologous positions
- Phylogeny reconstruction - Infer the tree using various methods
- Test reliability - Assess confidence in the estimated phylogeny
- Interpretation and application - Use the phylogeny to answer biological questions
Applications of Phylogenetics
Phylogenetic methods have diverse applications across biology:
- Inferring relationships among species and genes
- Estimating divergence times
- Identifying functional elements through comparative genomics
- Detecting molecular adaptation
- Forensics and paternity testing
- Studying the emergence and spread of viral pandemics
- Conservation biology and biodiversity assessment
2. Types and Anatomy of Phylogenetic Trees
Tree Representations

Phylogenetic trees can be drawn in various formats:
- Rectangular (dendogram): Branch lengths shown horizontally
- Circular: Taxa arranged in a circle
- Radial: Branches radiate from center
Bifurcating vs. Multifurcating Trees

- In a rooted tree: A node with more than 2 children
- In an unrooted tree: A node of degree 4 or greater
Polytomies can represent either uncertainty in relationships or rapid diversification events.
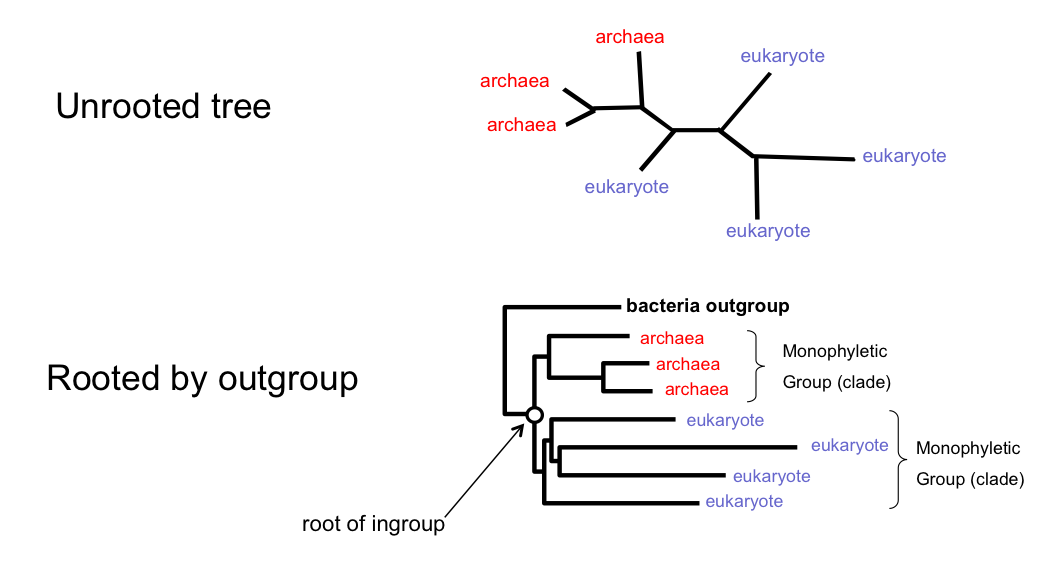
Rooted vs. Unrooted Trees

Key differences:
- Rooted trees show the direction of evolution and identify the common ancestor
- Unrooted trees show relationships but not evolutionary direction
- Many methods (parsimony, distance, likelihood without molecular clock) produce unrooted trees
Rooting Trees Using an Outgroup
Tree Anatomy
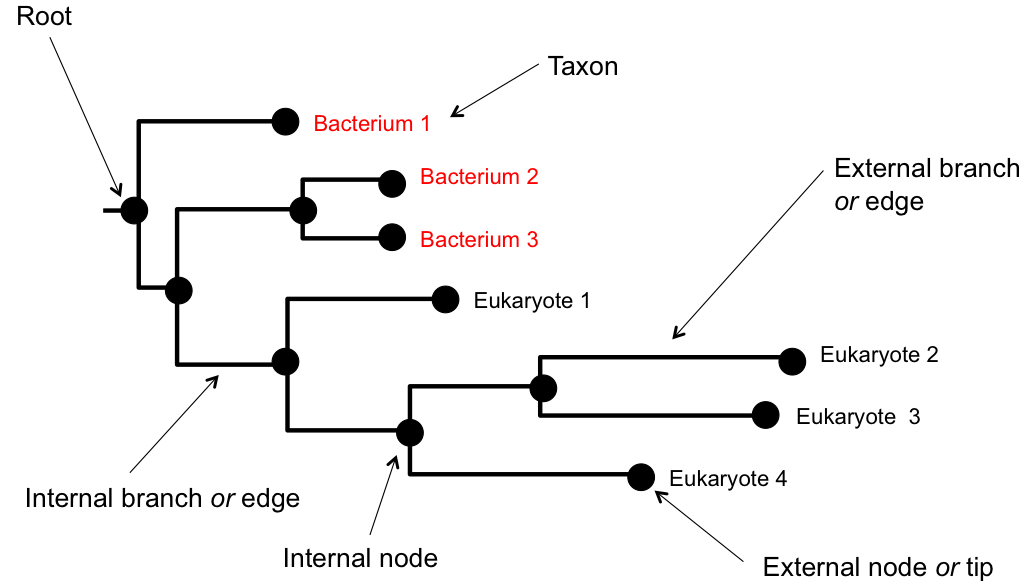
Essential tree terminology:
- Nodes: Points where branches meet (internal nodes = ancestors, tips/leaves = observed taxa)
- Branches/Edges: Lines connecting nodes (represent evolutionary lineages)
- Branch lengths: Can represent time or amount of evolutionary change
- Root: The common ancestor of all taxa in the tree
- Tips/Leaves: The observed taxa (species, genes, etc.)
3. The Universe of Possible Trees
How Many Trees Are There?
The number of possible tree topologies grows explosively with the number of taxa. For $n$ taxa, the number of:
Rooted, binary trees:
$$T_n^{(R)} = (2n-3)(2n-5)\cdots(3)(1) = \frac{(2n-3)!}{2^{n-2}(n-2)!}$$Tree Numbers in Perspective
| n (taxa) | # Rooted Trees | Context |
|---|---|---|
| 4 | 15 | Enumerable by hand |
| 5 | 105 | Enumerable by hand on a rainy day |
| 10 | 34,459,425 | ≈ Upper limit for exhaustive search |
| 20 | 8.2 × 10²¹ | ≈ Upper limit for branch-and-bound |
| 48 | 3.2 × 10⁷⁰ | ≈ Number of particles in the universe |
Counting Unrooted Trees
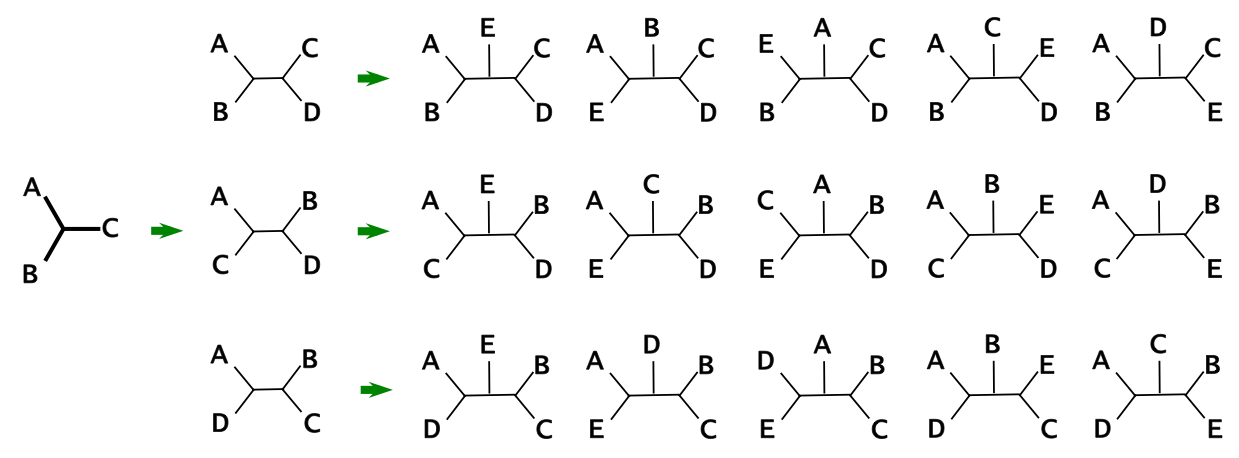
The number of unrooted trees can be calculated using stepwise addition:
- Start with 3 taxa: only 1 possible unrooted tree
- Each new taxon can be added to any branch
- Formula: $T_n = 1 × 3 × 5 × \cdots × (2n-5)$ for $n$ taxa
Measuring Tree Differences
The partition distance between two trees is the total number of bipartitions (splits) that are in one tree but not the other.
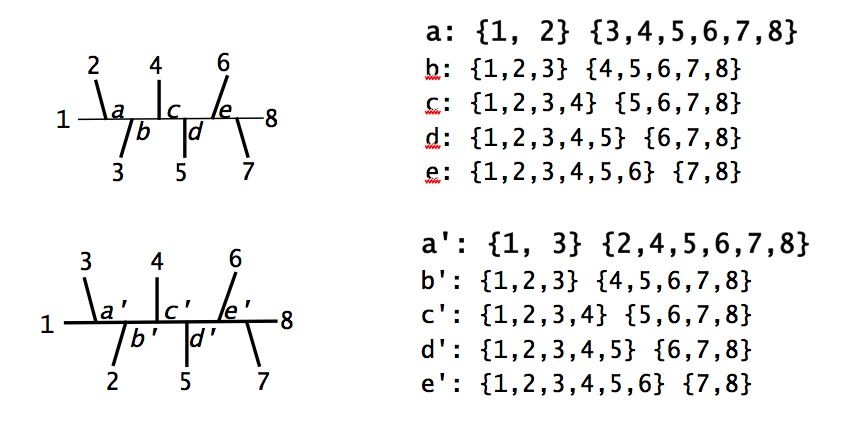
Properties of the Robinson-Foulds distance:
- Ranges from 0 (identical trees) to $2(n-3)$ for $n$ taxa
- Simple to compute but can be sensitive to small changes
- Treats all bipartitions equally (no weighting by branch length)
4. Overview of Phylogenetic Reconstruction
Types of Data
Phylogenetic reconstruction uses two main types of data:
Distance Data
Pairwise dissimilarities stored in a distance matrix
| A | B | C | D | E | |
|---|---|---|---|---|---|
| A | 0 | 3 | 5 | 6 | 5 |
| B | 3 | 0 | 4 | 7 | 6 |
| C | 5 | 4 | 0 | 5 | 4 |
| D | 6 | 7 | 5 | 0 | 1 |
| E | 5 | 6 | 4 | 1 | 0 |
Character Data
Discrete states for each taxon at each position
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
|---|---|---|---|---|---|---|---|---|---|
| A | 1 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 |
| B | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 |
| C | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 |
| D | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| E | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Reconstruction Approaches
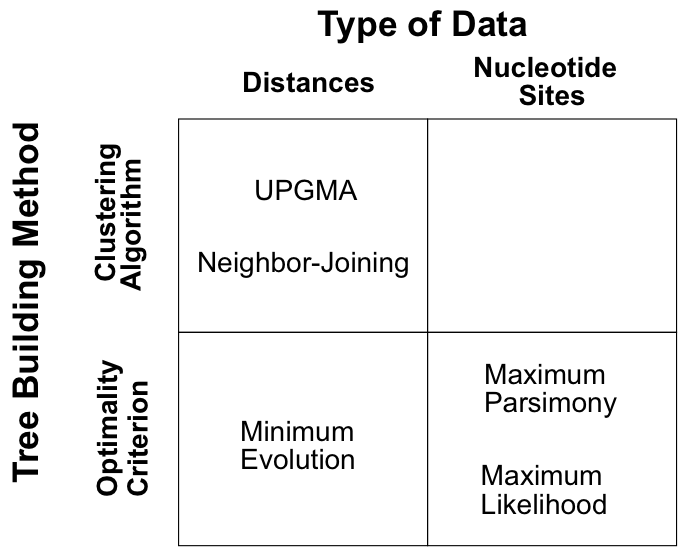
Given the enormous number of possible trees, we have three main strategies:
- Clustering algorithms: Build tree using a specific algorithm (fast but no optimality criterion)
- Optimality criteria: Define a score and find the tree(s) that optimize it
- Statistical inference: Find the most probable trees under an evolutionary model
5. Clustering Methods
Overview of Clustering Approaches
Clustering algorithms like UPGMA and Neighbor-Joining are:
- Usually very fast (polynomial time)
- Simple to implement and understand
- Based on greedy heuristics (make locally optimal choices)
- No explicit optimality criterion
- No measure of how good the tree is
- No information about alternative trees
UPGMA (Unweighted Pair Group Method with Arithmetic Mean)
UPGMA is one of the simplest tree-building methods:
UPGMA Algorithm
- Start with each taxon as a separate cluster
- Find the pair of clusters with smallest distance
- Join them into a new cluster
- Calculate distances from new cluster to all others using average distances
- Repeat until all taxa are joined
UPGMA Example
Step-by-step UPGMA
Initial distances:
| A | B | C | D | |
|---|---|---|---|---|
| A | 0 | 8 | 7 | 12 |
| B | 8 | 0 | 9 | 14 |
| C | 7 | 9 | 0 | 11 |
| D | 12 | 14 | 11 | 0 |
Step 1: Join A and C (smallest distance = 7)
New distances:
- $d_{B(AC)} = (d_{BA} + d_{BC})/2 = (8 + 9)/2 = 8.5$
- $d_{D(AC)} = (d_{DA} + d_{DC})/2 = (12 + 11)/2 = 11.5$

UPGMA Weaknesses
UPGMA can fail when evolutionary rates vary:

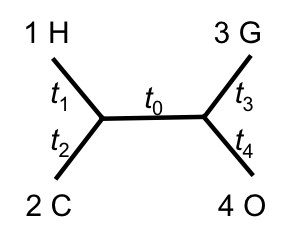
Neighbor-Joining (NJ) Algorithm
Saitou and Nei (1987) developed NJ to address UPGMA's limitations:
NJ corrects for unequal evolutionary rates by considering the average distance from each taxon to all others.
The NJ Algorithm
- Compute "average distance" for each taxon:
$$r_i = \frac{1}{n-2} \sum_j d_{ij}$$
- Calculate corrected distances:
$$d_{ij}^* = d_{ij} - r_i - r_j$$
- Join the pair with smallest $d_{ij}^*$
- Calculate branch lengths to new node:
$$d_{ik} = \frac{1}{2}(d_{ij} + r_i - r_j)$$
Time Complexity
Both UPGMA and NJ have $O(n^3)$ time complexity:
- $n$ steps (joining operations)
- Each step requires finding minimum distance: $O(n^2)$
- Updating distances: $O(n)$
6. Least Squares Distance Methods
Unlike clustering methods, least squares approaches have an explicit optimality criterion: minimize the difference between observed and tree-implied distances.
The Least Squares Criterion
Primate Example
| $d_{ij}$ | Human | Chimp | Gorilla | Orangutan |
|---|---|---|---|---|
| Human | 0 | 0.0965 | 0.1140 | 0.1849 |
| Chimp | 0 | 0.1180 | 0.2009 | |
| Gorilla | 0 | 0.1947 | ||
| Orangutan | 0 |
For any tree topology with branch lengths, we can calculate the sum of squared differences:
Where:
- $d_{ij}$ = observed distance between taxa $i$ and $j$
- $\hat{d}_{ij}$ = tree-implied distance (sum of branch lengths on path)
Optimization Process
For the primate example:
- 6 data points (pairwise distances)
- 5 free parameters (branch lengths)
- Use numerical optimization to minimize $S$
Results for Different Topologies
| Tree Topology | Least Squares Score (S) |
|---|---|
| ((Human,Chimp),Gorilla,Orangutan) | 0.000035 ✓ |
| ((Human,Gorilla),Chimp,Orangutan) | 0.000140 |
| ((Human,Orangutan),Chimp,Gorilla) | 0.000140 |

Advantages and Limitations
Advantages:
- Explicit optimality criterion
- Can compare different topologies
- Provides branch length estimates
Limitations:
- Still need to search tree space (NP-hard problem)
- Assumes distances are measured without error
- No statistical framework for uncertainty
Summary
This lecture introduced the fundamentals of phylogenetic analysis:
- Evolutionary information: Molecular sequences contain historical information that can be decoded through phylogenetic analysis
- Tree basics: Understanding tree anatomy, rooted vs. unrooted trees, and different representations
- Tree space: The number of possible trees grows explosively, making exhaustive searches impossible
- Distance methods:
- Clustering (UPGMA, NJ): Fast but no optimality measure
- Least squares: Explicit criterion but computationally intensive
- Key challenges: Accounting for rate variation, searching vast tree space, and quantifying uncertainty
- Higgs & Attwood (2005) "Bioinformatics and Molecular Evolution" - Sections 8.1, 8.3
- Yang (2006) "Computational Molecular Evolution" - Sections 3.1-3.3
- Durbin et al. (1998) "Biological Sequence Analysis" - Sections 7.1-7.3
- Saitou & Nei (1987) "The neighbor-joining method" - Mol Biol Evol 4:406-425
Check Your Understanding
- Why do unrooted trees contain less information than rooted trees?
- How does the number of possible trees change as you add more taxa?
- What assumption does UPGMA make that NJ does not?
- How do clustering methods differ from optimality-based methods?
- What information do we need to root an unrooted tree?
Phylogenetic Software
- MEGA: User-friendly GUI for distance and parsimony methods
- RAxML: Fast maximum likelihood phylogenetic inference
- IQ-TREE: Efficient phylogenetic software with model selection
- BEAST: Bayesian phylogenetic inference with molecular dating
- FigTree: Graphical viewer for phylogenetic trees