Sequence Homology
1. Introduction to Sequence Homology
Understanding homology is fundamental to phylogenetic analysis. When we observe similarity between sequences, we form hypotheses about their evolutionary relationships.
Key Concepts
- Homologous sequence regions are expected to display a degree of similarity
- The statement that two sequence regions are homologous is an evolutionary hypothesis based on similarity
- It is rarely possible to observe homology directly
- Homologous sequences are referred to as homologs
Types of Homologs
Different evolutionary events can lead to different types of homologous relationships:
- Orthology: Ancestral sequences separated by speciation
- Paralogy: Ancestral sequences separated by gene duplication
- Xenology: Homologs resulting from horizontal gene transfer
Examples from Nature
The globin family provides excellent examples of homologous proteins across diverse organisms:



2. Pairwise Alignment
The Goal of Pairwise Alignment
Aligning one sequence with another allows us to assess the homology between the two sequences.
Alignment serves several crucial purposes in sequence analysis:
- It breaks down the question of sequence similarity into smaller questions about character similarity
- It forms the basis for multiple sequence alignment
- It provides the foundation for phylogenetic reconstruction from molecular data
Dot Plots
A simple visual method for assessing pairwise homology. Consider these two sequences:
- "decided to build three ships, three Arks in space"
- "decided to build three Arks in space"
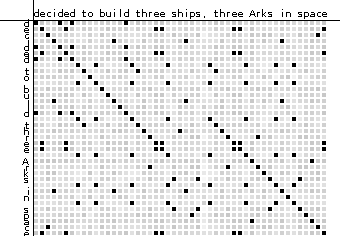
- Each row corresponds to a position on sequence 1
- Each column corresponds to a position on sequence 2
- Pixel (i,j) is colored if characters at site i on seq 1 and j on seq 2 match
- Diagonal lines indicate runs of matching sites
Scoring Alignments
Alignments are evaluated using scoring systems that assign numeric values to each column:
Column types and their typical scores:
- Identical: Positive score
- Conservative: Positive score
- Non-conservative: Negative score
- Gap: Negative score
Scoring Methods
Two main approaches are used for scoring alignments:
1. Model-based Scoring
Based on evolutionary models, particularly log-odds scoring:
Where:
- \(p_{ab}\) is the probability of observing residues a and b in homologous sequences
- \(f_a\) and \(f_b\) are the background frequencies of residues a and b
- \(\lambda\) is a scaling factor
2. Empirical Scoring
Based on observed substitution patterns in real sequences:
- PAM matrices
- BLOSUM matrices
- JTT matrix
- WAG matrix
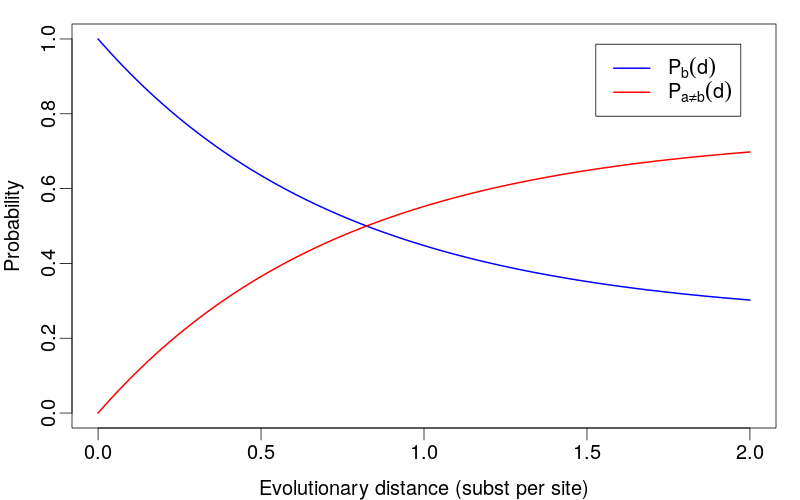
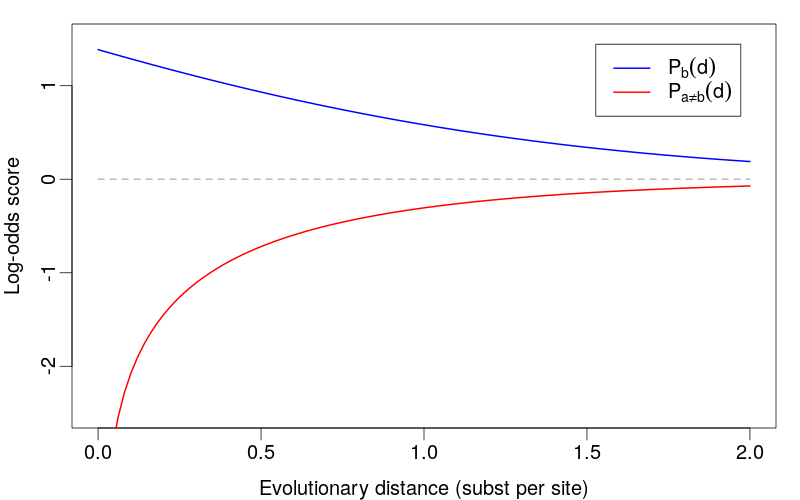
Evolutionary Interpretation
For homologous sequences with divergence time \(t\) and mutation rate \(\mu\):
Gap Penalties

Two main approaches for penalizing gaps in alignments:
- Linear gap penalty:
$$\gamma(g) = -gd$$where \(d\) is the gap penalty and \(g\) is the gap length
- Affine gap penalty:
$$\gamma(g) = -d - (g-1)e$$where \(d\) is the gap opening penalty and \(e\) is the gap extension penalty
3. Pairwise Alignment Algorithms
Dynamic Programming
Dynamic programming is a powerful method for solving combinatorial optimization problems. It guarantees finding the optimal solution and is based on the principle of optimality:
A sub-optimal solution of a sub-problem cannot be part of the optimal solution of the full problem.

Key features of dynamic programming:
- Computation is carried out from the bottom-up
- All solutions to sub-problems are stored in a table
- Each sub-problem is solved exactly once
- Only optimal solutions to sub-problems are used for the next level
Needleman-Wunsch Algorithm
The Needleman-Wunsch algorithm (1970) performs global alignment using dynamic programming.
Algorithm Overview
Given sequences X = (x₁, x₂, ..., xₘ) and Y = (y₁, y₂, ..., yₙ), where m and n are the lengths of sequences X and Y respectively, define F(i,j) as the score of the best alignment between the first i characters of X and the first j characters of Y.
Recurrence Relation
Initialization
- F(0,0) = 0
- F(i,0) = F(i-1,0) + s(xᵢ,-) for i > 0
- F(0,j) = F(0,j-1) + s(-,yⱼ) for j > 0
Biological Application
The Needleman-Wunsch algorithm is particularly useful when:
- Comparing full-length protein sequences from different species
- Aligning complete genes to study synteny
- Example: Aligning human and mouse insulin genes to study conservation
Computational Complexity
- Time complexity: O(mn) where m and n are sequence lengths
- Space complexity: O(mn) for the full matrix
- Traceback: O(m+n)
Interactive Demo
Try the Needleman-Wunsch algorithm with your own sequences:
Launch Interactive Demo →Smith-Waterman Algorithm
The Smith-Waterman algorithm computes local alignment, finding the best alignment of subsequences while ignoring scores of regions on either side.

Key Difference
The main difference from Needleman-Wunsch is the addition of a fourth option in the recurrence:
This allows the algorithm to:
- Start a new alignment at any position
- End an alignment when the score becomes negative
- Find multiple high-scoring local alignments
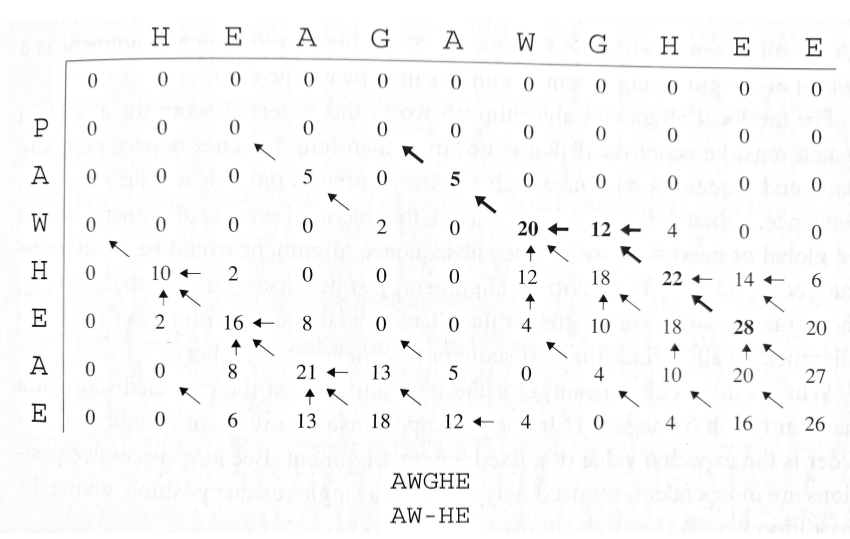
Specialized Variants
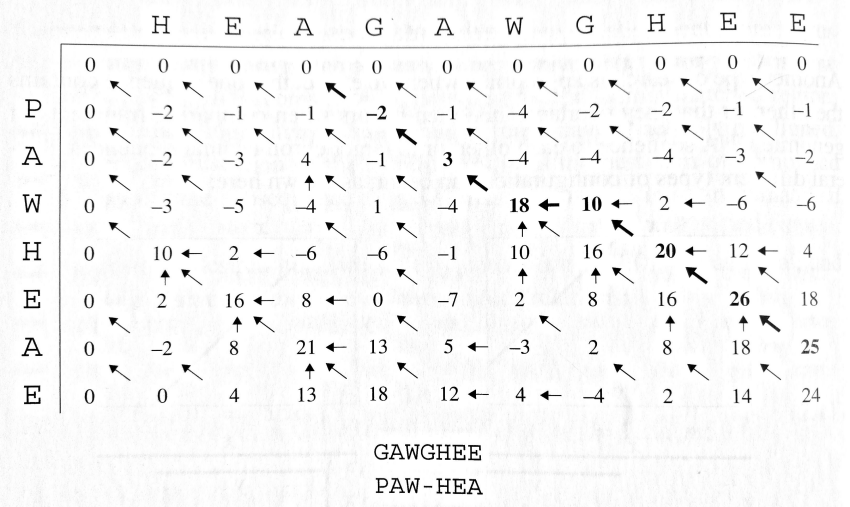
Repeated Matches
For finding multiple occurrences of a motif in a longer sequence:
- Useful for identifying repeated domains or motifs
- Asymmetric algorithm (short query vs. long target)
- Tracks multiple high-scoring regions above threshold T
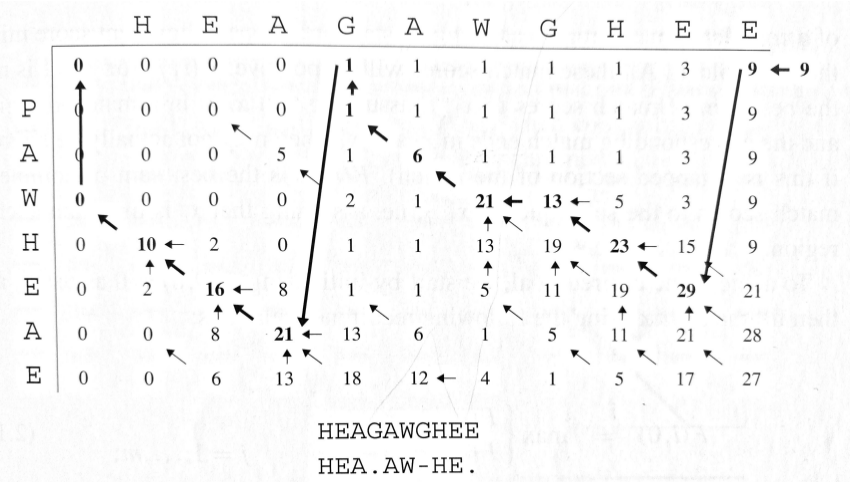
Overlap Matches
For sequence assembly and finding overlapping sequences:
- No penalty for overhanging ends: F(i,0) = F(0,j) = 0
- Useful in genome assembly projects
- Identifies sequences that overlap at their ends
Summary and Next Steps
In this lecture, we've covered the fundamental concepts of sequence homology and pairwise alignment:
- The definition and types of homology
- Methods for visualizing and scoring sequence alignments
- Dynamic programming algorithms for global and local alignment
- Specialized alignment variants for specific biological problems
These concepts form the foundation for:
- Multiple sequence alignment (next lecture)
- Phylogenetic tree construction
- Evolutionary rate estimation
- Functional annotation of sequences
- Durbin et al. (1998) "Biological Sequence Analysis" - Chapters 2-3
- Felsenstein (2004) "Inferring Phylogenies" - Chapter 11
- Original papers: Needleman & Wunsch (1970), Smith & Waterman (1981)