Likelihood and Substitution Models
1. Problems with Parsimony
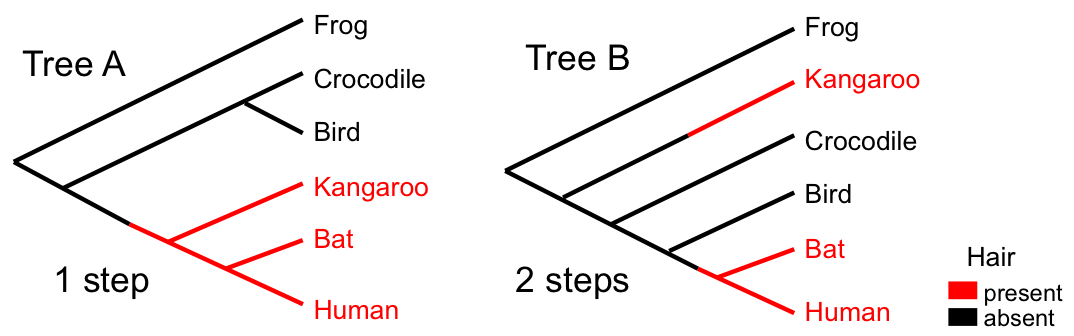
While parsimony is intuitive and computationally tractable for small problems, it has several limitations:
- "Large parsimony problem" is computationally expensive
- NP-hard problem requiring heuristic search algorithms
- No guarantee of finding global optimum
- Gives point estimates with no uncertainty quantification
- No confidence intervals or support values
- No way to compare alternative hypotheses statistically
- Questionable biological basis
- Assumes all changes are equally unlikely
- Ignores branch lengths
- Not model-based
- No formal hypothesis testing
- No model comparison
- Hidden problems
- Long branch attraction
- Inconsistency under certain conditions
2. Modelling Neutral Sequence Evolution
Why Use Models?
We need a model to relate what we observe (data) to what we want to know (hypotheses and parameters).
We use probabilistic models for molecular evolution because:
- We don't know enough about mutation mechanisms for deterministic models
- Stochastic effects are important at molecular level
- Allows uncertainty quantification
- Enables formal statistical inference
Genetic Distance: From p-distance to Model-based Estimates
The p-distance
The proportion of sites that differ between two aligned sequences.
Example: If 15 out of 100 sites differ, p-distance = 0.15
- Simple to calculate
- Always between 0 and 1
- Also called normalized Hamming distance
- BUT: underestimates true evolutionary distance (we'll see why)

p-distance Example

p-distance = 3/7 ≈ 0.43
Interpretation: 43% of sites differ between these sequences
Why p-distance Underestimates True Distance
The p-distance systematically underestimates the true genetic (evolutionary) distance because it cannot detect:

Biological Intuition
Imagine watching a bird feeder at different times:
- You see a korimako/bellbird at 9am and you again see a korimako/bellbird at 5pm.
- Same bird all day? Or did multiple birds use the spot?
- Without continuous observation, you can't tell!
Similarly, DNA sites may have changed multiple times between ancestors and descendants.
- Multiple substitutions: Same site changing more than once
- Parallel substitutions: Same change occurring independently
- Back-substitutions: Reversions to ancestral state
Relationship Between p-distance and Genetic Distance
Why does p-distance saturate at 0.75?
With 4 nucleotides and random substitutions:
- Probability of staying the same = 1/4
- Probability of being different = 3/4
- After infinite time, sequences become random with respect to each other
- Random sequences differ at 75% of sites
3. Continuous-Time Markov Chains (CTMCs)
Mathematical Framework
A stochastic process where:
- State X(t) is a function of continuous time
- Future states depend only on current state (Markov property)
- Transition rates are constant over time
What does this mean biologically?
A continuous-time Markov chain models how nucleotides change over time:
- Each site evolves independently
- Changes can happen at any time
- The probability of change depends only on current state, not history
- Like radioactive decay - constant probability of change per unit time
Example: An 'A' nucleotide doesn't "remember" how long it's been an 'A' - it always has the same probability of mutating in the next instant.
CTMCs obey the Chapman-Kolmogorov equation:
In plain English: To get from state X(t₀) to X(t₁), sum over all possible paths through intermediate states.
Relationship Between Q and P(t)
The transition probability matrix P(t) is the matrix exponential of Qt:
Intuition for the Matrix Exponential
This formula tells us:
- P(0) = I (no time = no change)
- P(small t) ≈ I + tQ (short time = approximately linear)
- P(large t) → stationary distribution (long time = equilibrium)
Think of it as compound interest for mutations!
CTMC Example
Two-State System
Consider a simplified DNA with only purines (R) and pyrimidines (Y):
This means:
- Rate R→Y: 2 per unit time
- Rate Y→R: 1 per unit time
Biological interpretation: Transitions (R↔Y) happen at different rates in each direction
Time spent in state before transition follows exponential distribution:
Time-Reversibility
A CTMC is time-reversible if the process looks the same running forward or backward in time. This is a mathematical convenience, not a biological claim — evolution clearly has a direction!
Why do we assume reversibility?
- It means we don't need to know the root position to calculate the likelihood of a tree (Felsenstein's "pulley principle")
- All standard substitution models (JC69, K80, HKY, GTR) are time-reversible
- Greatly simplifies computation
The formal mathematical definition (detailed balance equations) is given in Appendix A.3.
4. DNA Substitution Models
Now let's see how CTMCs are used to model DNA evolution. We'll start simple and build complexity.
Jukes-Cantor (JC69) Model
The Simplest Model

Assumptions:
- All substitutions equally likely
- Equal base frequencies (25% each)
- One parameter: μ (overall rate)
Rate matrix:
Transition probabilities:
Genetic Distance Under JC69
Given observed p-distance, we can estimate the true genetic distance:
- Formula undefined if p ≥ 3/4 (sequences too divergent)
- Assumes all changes equally likely (unrealistic)
- Assumes equal base composition (often violated)
Kimura 2-Parameter (K80) Model
Adding Biological Realism

Key innovation: Transitions ≠ Transversions
- Transitions (α): Purine↔Purine, Pyrimidine↔Pyrimidine
- Transversions (β): Purine↔Pyrimidine
- Usually α > β (transitions more common)
Rate matrix:
Why are transitions more common?
Chemical similarity:
- Purines (A,G): Two-ring structures
- Pyrimidines (C,T): One-ring structures
- Replacing like-with-like causes less structural disruption
HKY Model
Hasegawa-Kishino-Yano (1985)

Features:
- Different transition/transversion rates
- Unequal base frequencies (πA, πC, πG, πT)
- Reflects GC-content variation
Rate matrix:
General Time Reversible (GTR) Model
The Most General Model

Features:
- All substitution types can have different rates
- 6 rate parameters + 4 frequency parameters
- Most flexible time-reversible model
- Includes all previous models as special cases
Rate matrix:
Model Comparison Summary
| Model | Parameters | Key Feature | When to Use |
|---|---|---|---|
| JC69 | 1 | All equal | Very similar sequences |
| K80 | 2 | Ts/Tv ratio | Moderate divergence |
| HKY | 5 | +Base frequencies | GC-content varies |
| GTR | 9 | All different | Complex datasets |
Rate Heterogeneity Among Sites
Biological Reality
Not all sites in a gene evolve at the same rate:
- Active sites: Highly conserved (slow evolution)
- Structural regions: Moderate constraints
- Surface loops: Fewer constraints (fast evolution)
- Synonymous sites: Often evolve fastest
We model rate variation using a gamma distribution with a single shape parameter α (see Appendix A.5 for the formula):

Understanding the shape parameter α
- α < 1: Most sites evolve slowly, few evolve very fast
- α = 1: Exponential distribution
- α > 1: Bell-shaped distribution
- α → ∞: All sites evolve at same rate
In practice, the continuous gamma distribution is approximated by a discrete gamma with \(k\) rate categories (typically \(k=4\)), each with equal probability \(1/k\). This is the "+Γ" in model names like "GTR+Γ".
Model Comparison Example
HIV Distance Estimates
HIV-1B vs other strains (env gene):
| Comparison | p-distance | JC69 | K80 | Tajima-Nei |
|---|---|---|---|---|
| HIV-O | 0.391 | 0.552 | 0.560 | 0.572 |
| SIVcpz | 0.266 | 0.337 | 0.340 | 0.427 |
| HIV-1C | 0.163 | 0.184 | 0.187 | 0.189 |
Observation: Model choice matters more for divergent sequences!
5. Likelihood-Based Phylogenetic Inference
From Distances to Likelihood
Distance methods lose information by summarizing sequences as pairwise distances. Likelihood methods use all the data.
The Likelihood Function
The likelihood for parameter θ under model M given data D is:
In words: "The probability of observing our data if the parameter values were θ"
Important: The likelihood is NOT a probability distribution over θ!
Simple Example: Coin Flips
5 tosses give: D = (H,T,T,H,T)
What's the probability of getting exactly this sequence?
This is the likelihood function L(f|D)!
Maximum Likelihood Inference
The maximum likelihood estimate is the parameter value that makes the observed data most probable.
Why Maximum Likelihood?
- Intuitive: Choose parameters that make data most likely
- Optimal properties: Consistent, efficient (with enough data)
- General framework: Works for any statistical model
- BUT: Only gives point estimates, no uncertainty!
Tree Likelihood
For phylogenetics, we want P(alignment | tree, model):

The likelihood calculation involves:
- For each site in the alignment:
- Consider all possible ancestral states
- Calculate probability of observing tip states
- Sum over all possibilities
- Multiply across sites (assumes independence)
Felsenstein's Pruning Algorithm
Joseph Felsenstein (1973) solved this using dynamic programming:
The Pruning Algorithm
Key idea: Work from tips toward root, storing partial likelihoods
Define: Lk(s) = likelihood of subtree below node k if node k has state s
For leaf nodes:
For internal nodes with children i and j:
Where P(x|s,t) is the probability of state s changing to x in time t
Why This Works
Instead of considering all 4^(n-1) combinations:
- Store 4 numbers at each node (partial likelihoods)
- Build up from tips to root
- Reuse calculations (dynamic programming principle)
Result: Linear time in number of taxa!
Maximum Likelihood Software
ML Phylogenetic Software
- IQ-TREE (Nguyen et al., 2015)
- State-of-the-art tree search
- Automatic model selection
- Ultrafast bootstrap
- RAxML (Stamatakis, 2014)
- Highly optimized for large datasets
- Excellent parallelization
- Standard in many pipelines
- PhyML (Guindon et al., 2010)
- User-friendly
- Good for moderate datasets
- Fast approximate likelihood ratio tests
- FastTree (Price et al., 2009)
- Approximate but very fast
- Can handle millions of sequences
- Good for initial analyses
Limitations of Maximum Likelihood
- No confidence intervals
- How certain are we about the tree?
- Bootstrap provides some help (but it's ad hoc)
- Model selection challenges
- How do we choose between JC69, K80, HKY, GTR?
- Likelihood ratio tests only work for nested models
- Can't incorporate prior knowledge
- What if we know some clades are unlikely?
- What if we have fossil calibrations?
We need a framework that handles uncertainty properly → Bayesian inference!
Mathematical Appendix
A.1 Master Equation / Kolmogorov Forward Equation
The Chapman-Kolmogorov equation for a CTMC can be written in differential form as the master equation (also called the Kolmogorov forward equation):
where \(Q\) is the instantaneous rate matrix with off-diagonal elements \(Q_{ij}\) giving the rate from state \(i\) to \(j\), and diagonal elements \(Q_{ii} = -\sum_{j \neq i} Q_{ij}\) ensuring rows sum to zero. Because this equation is linear in the probabilities, its solution is the matrix exponential: \(\vec{p}(t) = \exp(Qt)\,\vec{p}(0)\).
See Decoding Genomes §5.2.2.3.
A.2 Matrix Exponential
The matrix exponential is defined by the Taylor series:
Useful properties:
- \(\exp(\mathbf{0}) = I\)
- When \(AB = BA\): \(\exp(A+B) = \exp(A)\exp(B)\)
- If \(A = \text{diag}(a_1,\ldots,a_n)\), then \(\exp(A) = \text{diag}(e^{a_1},\ldots,e^{a_n})\)
Worked Example: 2-state system
For the rate matrix \(Q = \begin{bmatrix} -1 & 1 \\ 0.6 & -0.6 \end{bmatrix}\):
- At \(t=1\): \(P(1) = \begin{bmatrix} 0.501 & 0.499 \\ 0.299 & 0.701 \end{bmatrix}\)
- At \(t=2\): \(P(2) = \begin{bmatrix} 0.400 & 0.600 \\ 0.360 & 0.640 \end{bmatrix}\)
- As \(t \to \infty\): \(P(\infty) = \begin{bmatrix} 0.375 & 0.625 \\ 0.375 & 0.625 \end{bmatrix}\)
Note how rows converge to the stationary distribution \(\pi = [0.375, 0.625]\): the starting state no longer matters.
See Decoding Genomes §5.2.2.
A.3 Detailed Balance and Time-Reversibility
A CTMC is time-reversible if and only if its stationary distribution \(\pi\) satisfies the detailed balance condition:
Equivalently, there exists a row vector \(\Pi\) of state probabilities such that \(\Pi Q = 0\).
Verifying reversibility for the 2-state example
For \(Q = \begin{bmatrix} -2 & 2 \\ 1 & -1 \end{bmatrix}\) with \(\pi = [1/3, 2/3]\):
- \(\pi_0 Q_{01} = \frac{1}{3} \times 2 = \frac{2}{3}\)
- \(\pi_1 Q_{10} = \frac{2}{3} \times 1 = \frac{2}{3}\) ✓
Detailed balance is satisfied, so this CTMC is time-reversible.
See Decoding Genomes §5.2.3.
A.4 Deriving JC69 Genetic Distance
Under the Jukes-Cantor model, the probability that a site differs after time \(t\) is:
Setting \(p_{\text{diff}}\) equal to the observed \(p\)-distance and solving for \(\mu t\) gives the JC69 distance estimator:
See Decoding Genomes §5.3.
A.5 Gamma Distribution and JC69+Gamma Distance
The gamma distribution with shape parameter \(\alpha\) (and rate parameter \(\alpha\), so that the mean rate is 1) has probability density:
When site rates are gamma-distributed, the JC69 distance formula generalises to:
See Decoding Genomes §5.4.
A.6 Maximum Likelihood Estimation: Derivation
For the coin-flip example with \(n\) heads in \(N\) flips:
Taking the derivative and setting it to zero:
Solving gives \(\hat{f} = n/N\), the observed proportion of heads.
See Decoding Genomes §5.5.
Summary
This lecture introduced model-based phylogenetic inference:
- Why models?
- Parsimony has serious limitations
- Models enable statistical inference
- Can test hypotheses formally
- Substitution models as CTMCs:
- Continuous-time Markov chains model sequence evolution
- Models range from simple (JC69) to complex (GTR+Γ)
- More complex models fit data better but risk overfitting
- Key biological insights:
- p-distance underestimates true distance due to multiple hits
- Transitions more common than transversions
- Base composition varies across genomes
- Different sites evolve at different rates
- Likelihood methods:
- Use all information in the alignment
- Felsenstein's algorithm makes computation feasible
- BUT: still only point estimates
- Next step: Bayesian inference provides a complete framework for uncertainty
Key Takeaways
- For biologists: Models let us correct for unseen changes and test evolutionary hypotheses
- For computer scientists: Phylogenetics offers rich algorithmic challenges (DP, matrix exponentials, tree search)
- For everyone: Understanding models is crucial for interpreting phylogenetic results
- Stadler et al. (2024) Decoding Genomes - Chapter 5: Molecular Evolution, Ch 6.3.3 (maximum likelihood methods)
- Felsenstein (2004) "Inferring Phylogenies" - Chapters 13-16
- Yang (2014) "Molecular Evolution: A Statistical Approach" - Chapters 1-2
- Drummond & Bouckaert (2015) "Bayesian Evolutionary Analysis with BEAST" - Chapter 2
Check Your Understanding
- Why does p-distance underestimate true evolutionary distance?
- What biological reasons explain why transitions are more common than transversions?
- How does Felsenstein's algorithm achieve computational efficiency?
- What are the key differences between JC69, K80, HKY, and GTR models?
- Why isn't likelihood a probability distribution over parameters?