Characters
- An organism is comprised of a set of features.
- When organisms/taxa differ with respect to a feature (e.g. presence/absence of wings or a different nucleotide at a specific sites in a sequence) the different conditions are termed character states.
- The collection of character states (e.g. A, C, G, T) with respect to a feature constitute a character.
- Similarities and differences in character states provide the basis for inferring phylogeny (i.e. provide evidence of ancestral/evolutionary relationships).
When do characters support the correct tree?
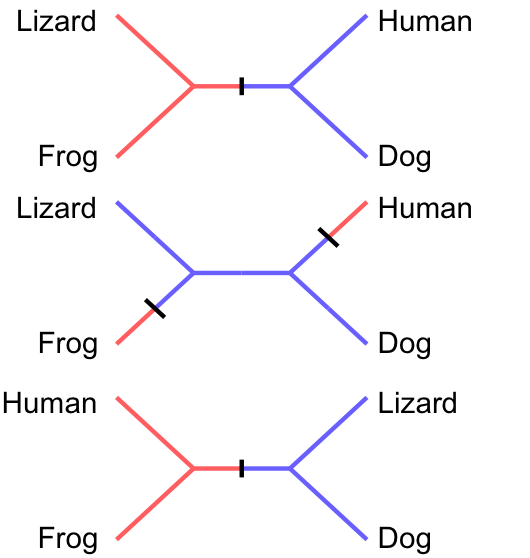
Hair: — absent — present
Congruence: Unique and unreversed character
Tail (adult): — absent — present
Homoplasy: Character evolved independently
Tail (adult): — absent — present
Parsimony gives incorrect inference
Reversals
- If a character reverts to an ancestral state this can also affect phylogenetic inference.
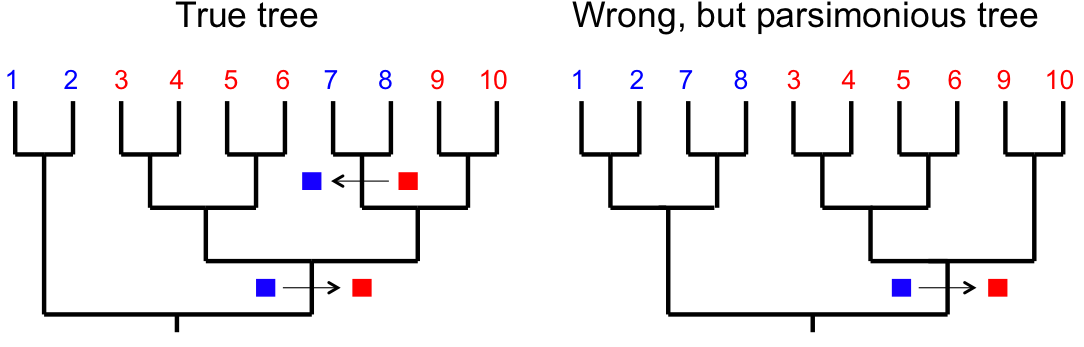
True tree: one change (■) + one reversal (■). Wrong tree: two independent changes — appears more parsimonious (fewer total events).
Parsimony
- Key issue: how to separate homoplasy from congruence
- The parsimony criterion favors hypotheses that maximize congruence and minimise homplasy
- Parsimony methods provide one way of choosing among alternative phylogenetic hypotheses
- It depends on the idea of the fit of a character to a tree
- Initially, we can define the fit of a character to a tree as the minimum number of steps required to explain the observed distribution of character states at the tips.
- This is an optimization problem
- Characters differ in their fit to different trees
Parsimony
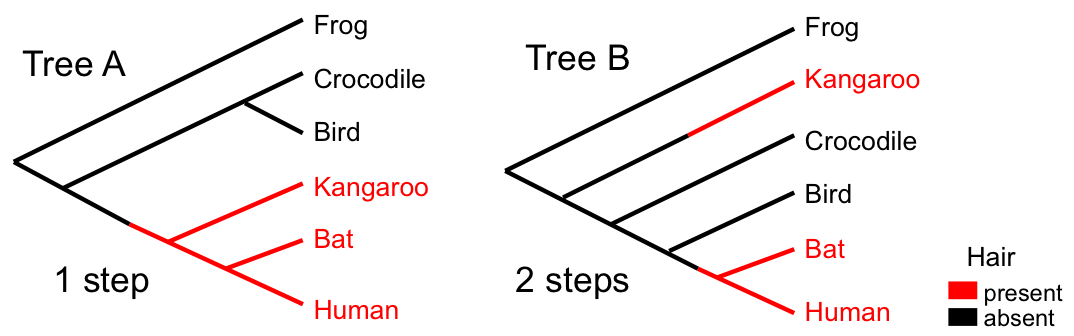
Tree A requires 1 step (gain of hair). Tree B requires 2 steps. Parsimony prefers Tree A.
- Given a set of characters, such as aligned sequences, parsimony analysis works by determining the fit (minimum number of steps) of each character on a given tree.
- The sum over all characters is called the tree length.
- The most parsimonious trees (MPTs) have the minimum tree length.
Parsimony informative sites
- Some sites (columns) have the same score on every tree.
- For example, a site with all bases the same will always score zero, regardless of the tree.
- Or a site with all bases the same except one will always score one.
- To get different scores on different trees, need at least two characters each of which appear in at least 2 taxa.
Finding optimal trees
- Exhaustive tree search evaluates all possible trees
- Branch-and-bound
- Heuristic search is not guaranteed to find the optimal tree
- stepwise addition
- star decomposition
- branch swapping
- nearest neighbor interchange (NNI)
- subtree-pruning and regrafting (SPR)
- tree bisection and reconnection (TBR)
Branch and bound example
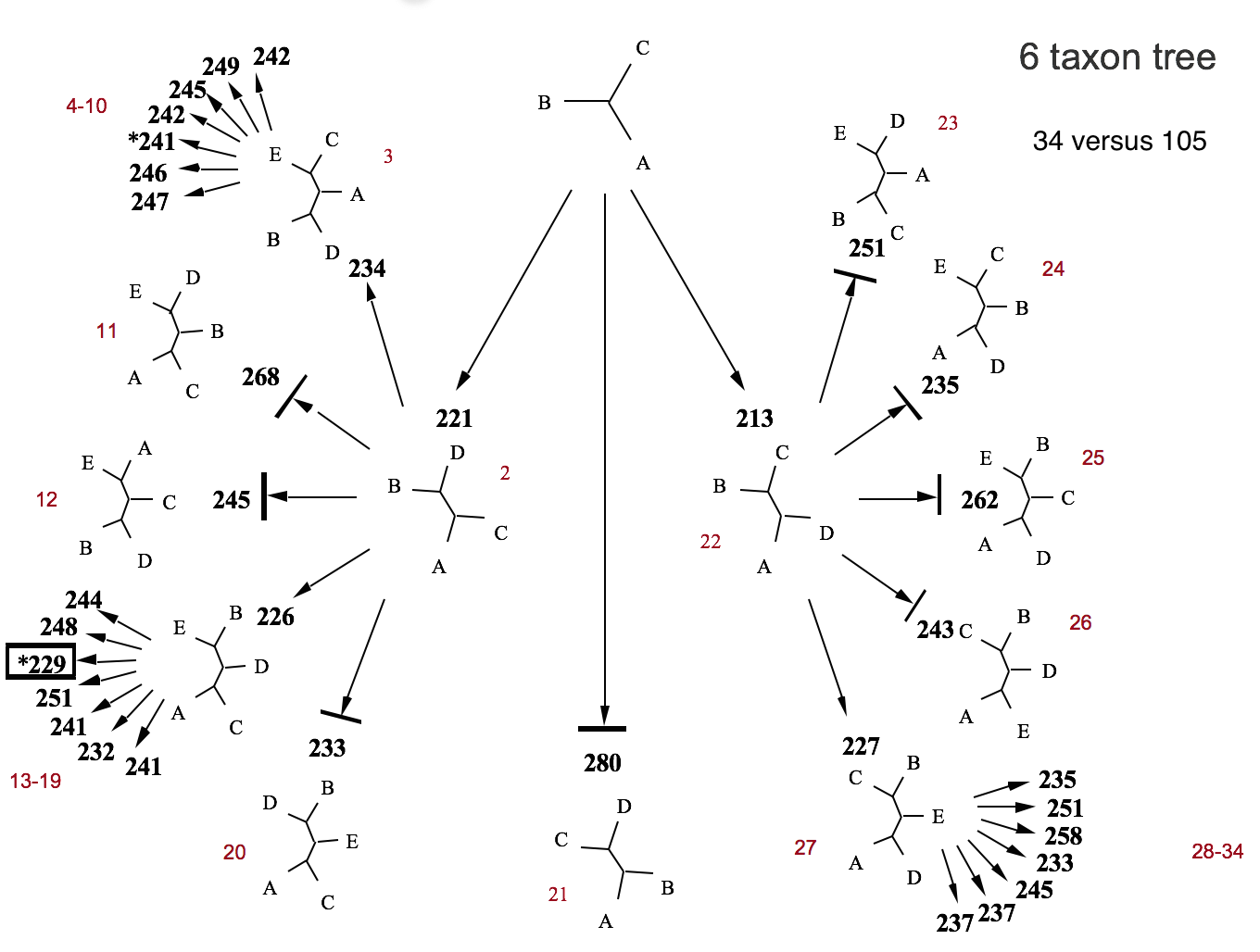
Branch-and-bound search for 6 taxa. Numbers show parsimony scores. The optimal score (*229) prunes branches with scores exceeding the current best. Searches 34 of 105 possible trees.
Heuristics
- The number of possible trees increases exponentially with the number of taxa making exhaustive searches impractical for many data sets (an NP complete problem)
- Heuristic methods are used to search tree space for most parsimonious trees by building or selecting an initial tree and swapping branches to search for better ones
- The trees found are not guaranteed to be the most parsimonious - they are best guesses
- General approach is starting tree + local search
- Starting tree constructed by adding one sequence at a time, perhaps with some searching in between additions
- More on this in later lectures
Representing trees
| Node | Left | Right | Taxon |
| 1 | 2 | 7 | |
| 2 | 3 | 6 | |
| 3 | 4 | 5 | |
| 4 | null | null | A |
| 5 | null | null | B |
| 6 | null | null | C |
| 7 | 8 | 9 | |
| 8 | null | null | D |
| 9 | null | null | E |
Fitch Parsimony
| Node | Left | Right | Taxon | States | Mark |
| 1 | 2 | 7 | |||
| 2 | 3 | 6 | |||
| 3 | 4 | 5 | |||
| 4 | null | null | A | ||
| 5 | null | null | B | ||
| 6 | null | null | C | ||
| 7 | 8 | 9 | |||
| 8 | null | null | D | ||
| 9 | null | null | E |
For internal nodes $v$: $L$ = left child, $R$ = right child.
If $\text{states}(L) \cap \text{states}(R) \neq \emptyset$: $\text{states}(v) \leftarrow \text{states}(L) \cap \text{states}(R)$
Else: $\text{states}(v) \leftarrow \text{states}(L) \cup \text{states}(R)$; Make a mark.
Leaves $v$: $\text{states}(v) \leftarrow$ state for that taxon.
Length = number of marks
Worked example: Fitch parsimony
Click leaf states to edit. Click internal states for detail.
Weighted parsimony (Sankoff algorithm)
| Node | Left | Right | Taxon | m(A) | m(C) | m(G) | m(T) |
| 1 | 2 | 7 | |||||
| 2 | 3 | 6 | |||||
| 3 | 4 | 5 | |||||
| 4 | null | null | A | ||||
| 5 | null | null | B | ||||
| 6 | null | null | C | ||||
| 7 | 8 | 9 | |||||
| 8 | null | null | D | ||||
| 9 | null | null | E |
| A | C | G | T | |
| A | 0 | 2 | 1 | 2 |
| C | 2 | 0 | 2 | 1 |
| G | 1 | 2 | 0 | 2 |
| T | 2 | 1 | 2 | 0 |
Internal nodes (L = left child, R = right child): $m[v,X]=\underset{Y}{\min}\{m[L,Y]+c(X,Y)\}+\underset{Z}{\min}\{m[R,Z]+c(X,Z)\}$
Leaves: $m[v,X] = \begin{cases} 0 & \text{if character state for } v \text{ is } X\\ \infty & \text{otherwise}\end{cases}$
Worked example: weighted parsimony
Character states: A=A, B=C, C=C, D=A, E=C. Click any cell to see computation.
Complexity of calculating tree length for a single tree
- Given n taxa there are $2n − 1$ nodes in a rooted binary tree,
- If there are S possible character states (S = 4 for DNA) then at each node we need to do $O(S^2)$ calculations
- If the sequence alignment is L long, then there are L characters to consider.
$O(nS^{2}L)$ time
$O(nS)$ space
Parsimony summary
- The “small parsimony problem” is efficiently computed by dynamic programming on the tree
- The “large parsimony problem” requires computing the parsimony score on all trees - there is no efficient solution for this
- The maximum parsimony principle attempts to find the evolutionary tree that requires the least number of events to explain the data.
- It is not based on a model, and does not allow for the possibility that multiple substitutions may have occurred on a branch in the tree.
Recommended Reading
- Chapter 6.3.2: Parsimony method
- 6.3.2.1 Parsimony score example
- 6.3.2.3 Fitch algorithm
- 6.3.2.5 Time complexity
- 6.3.2.6 Statistical inconsistency