The Phylogenetic Likelihood
$$\Pr(D|T, \mu)$$
- Computed by Felsenstein's pruning algorithm
- $D$ is a multiple sequence alignment of $n$ sequences,
- $T$ is a phylogenetic tree with $n$ leaves, and
- $\mu$ are the parameters of the chosen CTMC-based model of sequence evolution.
- Just a product of site pattern probabilities: note the obvious generalizations to multiple alignments that share a tree, or to phylogenetic networks that allow different sites to evolve under distinct "local" trees.
We're Bayesians: we need a probability distribution for $T$!
The Phylogenetic Posterior
Standard application of Bayes theorem gives the posterior:
$$P(T,\mu,\theta|D) = \frac{\Pr(D|T,\mu, \theta)P(T,\mu,\theta)}{\Pr(D)}$$But most phylogenetic models assume that $\theta$ only effects the probability of the data via the tree $T$, and likewise that $\mu$ has no effect on the tree branching process, leading to:
$$P(T,\mu,\theta|D) = \frac{1}{\Pr(D)}\Pr(D|T,\mu)P(T|\theta)P(\mu,\theta)$$
- $P(T|\theta)$ is the "tree prior" parameterized by $\theta$.
- $P(\mu,\theta)$ are the parameter priors.
- What is $\Pr(D)$?
- Is the tree prior really a prior? (i.e. does it depend on the data?)
The Neutrality Assumption
Because of the way we've factorized the joint probability for the data and model parameters, we are implicitly assuming that our alignment could have been produced in the following fashion:



Separating the process of tree generation from that of sequence evolution implies neutrality.
Tree Priors
- Tree priors allow us to specify a generative model for the genealogy.
- While this model may not involve genetic evolution, it may depend on speciation rates, population sizes, migration rates, etc. etc.
- A huge fraction of phylogenetics inference research is focused on developing and efficiently implementing tree priors!

The Yule model for speciation
- Simple model for speciation due to Yule (1924).
- Constant rate branching process where branching occurs uniformly across extant lineages at a rate of $\lambda$ per lineage per unit time.
- Number of lineages $k$ evolves under the following master equation: $$\dot{p}(k,t) = \lambda (k-1)p(k-1,t) - \lambda k p(k,t)$$
- Write this in chemical kinetics formalism: $X \overset{\lambda}{\longrightarrow} 2X$

Birth-death(-sampling) priors
- Most of the recent groundwork on this is due to Tanja Stadler (ETH, Zürich).
- Generalization of Yule process to account for extinction and sampling.
- Linear speciation, extinction and sampling rates.
- Prior is over sampled trees: assumes unobserved full tree.

Coalescent priors
- Original theory due to Kingman (1980).
- Model the shape of gene trees within a species.
- Can be obtained as the limit of a number of population genetic models, including the Wright-Fisher model and the Moran model.
- Provide an important link between tree shape and population size!

Multi-species coalescent priors
- Hierarchical models that embed gene trees within species trees.
- Species tree described using a birth-death prior.
- Gene trees described by coalescent priors for populations which subdivide at speciation events.
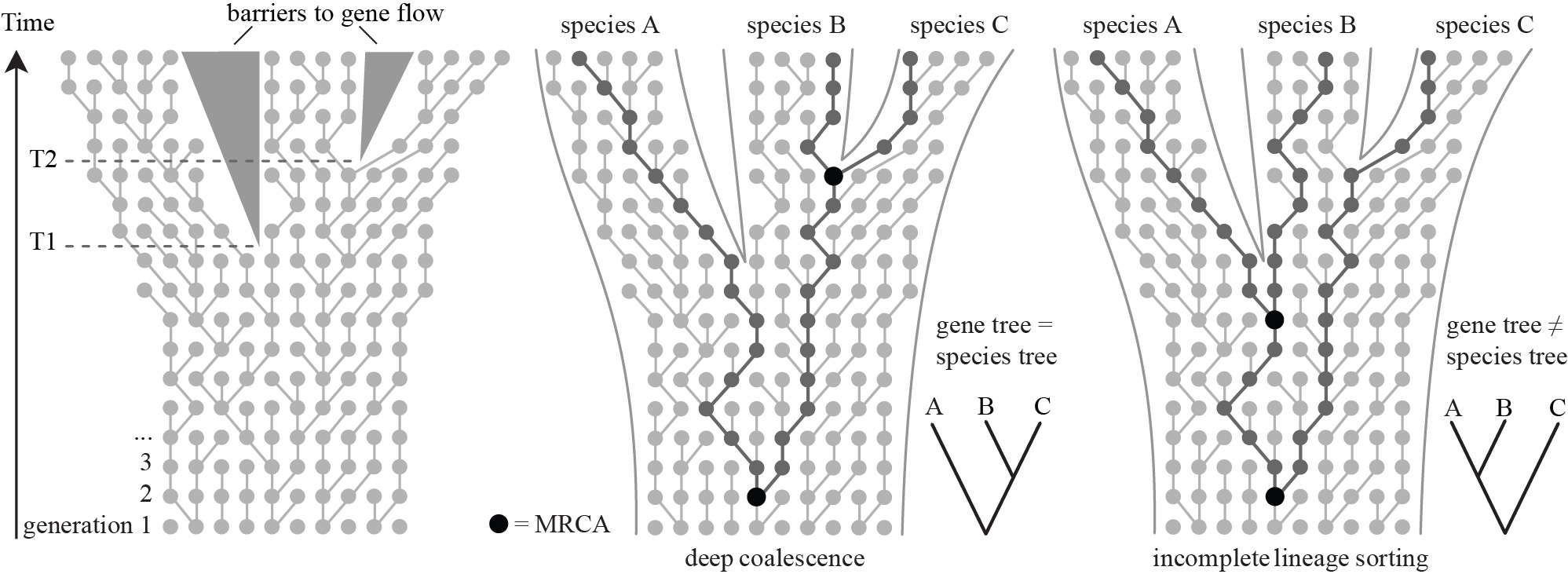
MCMC in Tree Space
Basic MCMC Theory
Think of MCMC like exploring a fitness landscape: current position = current tree & parameters, proposed moves = small "mutations", acceptance/rejection = selection. We don't need the whole landscape—just the relative heights of where we are vs. where we're going.
MCMC works by simulating a stochastic trajectory according to: $$p_{i+1}(x)=\int p_i(x')W(x|x')\mathrm{d}x'$$ where $$W(x'|x) = q(x'|x)\alpha(x'|x) + \left(1-\int q(x''|x)\alpha(x''|x)\mathrm{d}x''\right)\delta(x'-x)$$
A sufficient (but unnecessary!) condition for $\pi(x)$ to be the equilibrium distribution is detailed ballance, which implies:
$$\pi(x')q(x|x')\alpha(x|x')=\pi(x)q(x'|x)\alpha(x'|x)$$which is satisfied by the following acceptance probability:
$$\alpha(x'|x)=\min\left[1, \frac{\pi(x')}{\pi(x)}\times\frac{q(x|x')}{q(x'|x)}\right]$$
Basic MCMC Theory (continued)
$$\alpha(x'|x)=\min\left[1, \frac{\pi(x')}{\pi(x)}\times\frac{q(x|x')}{q(x'|x)}\right]$$
- By tuning the acceptance probability, any probability distribution can be targeted.
- Only the ratio $\pi(x')/\pi(x)$ appears: normalization need not be known.
- The only requirement for $q(x'|x)$ is that it produce random walks that explore the entire state space for which $\pi(x)$ has support (cf. irreducibility).
- For high-dimensional state spaces, $W(x'|x)$ is usually decomposed into several distinct transitions: $W(x'|x)=\sum_j W_j(x'|x)$ that each modify some small part of the state and individually satisfy detailed balance in the steady state.
- The ratio $q(x|x')/q(x'|x)$ is called the Hastings Ratio and accounts for any asymmetry in the proposal rates between $x$ and $x'$.
Proposal distributions/operators for Trees
Need to identify a set of proposals $q_j(x'|x)$ when $x$ is a point in the space of rooted time trees.

Stopping criteria
- How can we tell when a phylogenetic MCMC calculation has reached equilibrium?
- How do we know when we've collected enough samples?
- One approach is to compute ESS (using empirical estimates of autocorrelation time) for each
parameter and a number of tree summary statistics (e.g. tree height and tree "length").
- Assume that once all of these scores are sufficiently high (e.g. > 200) we have adequately sampled the posterior.
- Examine output of several independent (and independently
initialized chains. A necessary (not sufficient) condition for
convergence is that sample distributions converge as the chains
converge to equilibrium.
- Always do this!
Are we there yet?
The AWTY application applies a number of different statistics to assess the convergence of the tree state. It relies heavily on comparing the result of multiple runs.
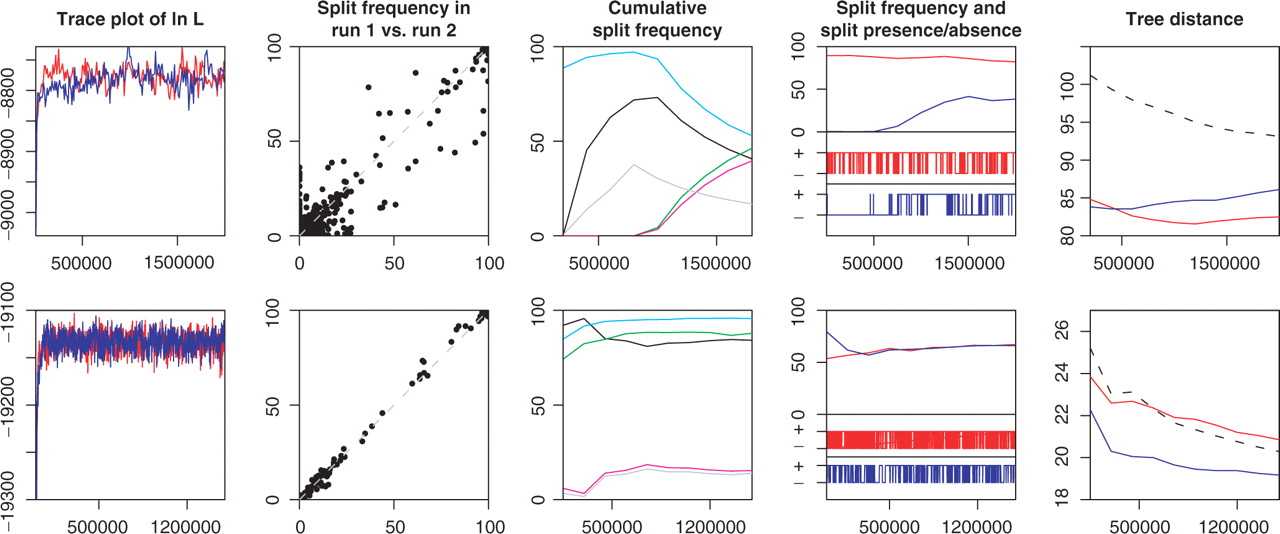
Post-processing
Parameter samples
Logs of individual parameters can be considered samples from the marginal posteriors of those parameters.
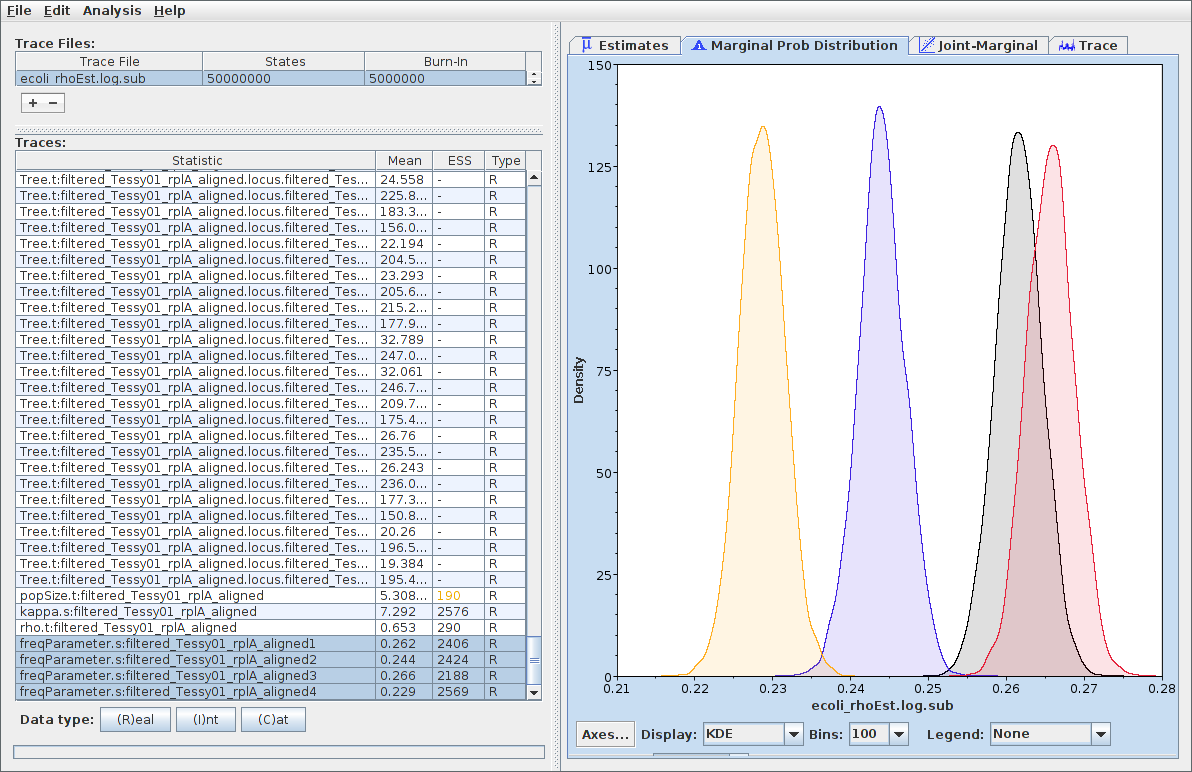
Tree samples
- Summary statistics (e.g. age of the MRCA of a pair of taxa) can be computed and their marginal distributions assessed.
- A number of different approaches can be used to produce a "summary" tree meant to represent the best guess at the true tree. (None of these algorithms are perfect!!)
- Strict consensus
- Include only clades that appear in ALL sampled trees.
- Majority rule consensus
- Include clades that appear in >50% of the sampled trees.
- Maximum clade credibility tree
- The sampled topology for which the product of the posterior clade probabilities is maximized.
In BEAST, summary trees are produced using the MCC tree method via the program TreeAnnotator.
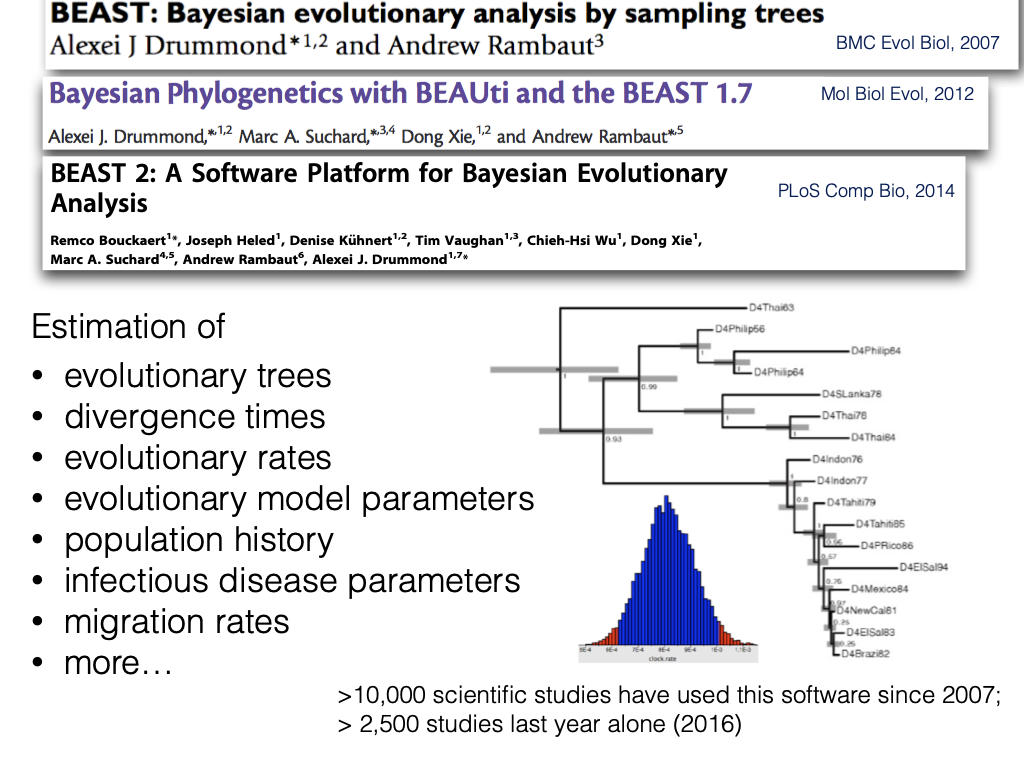
Bayesian Phylogenetic Inference Software
Popular Bayesian phyogenetic inference software:
- MrBayes (Huelsenbeck and Ronquist, Bioinformatics, 2001)
- Early CLI program for phylogenetic inference.
- RevBayes (Höhna et al., Syst. Biol., 2016)
- R-like syntax for specifying phylogenetic models.
- BEAST/BEAST2 (Drummond and Rambaut, 2007; Bouckaert et al., 2014)
- XML specification of phylogenetic models. Extensible.
Some software implementing special models:
- MIGRATE
- Performs inference under the Structured Coalescent. (i.e. subpopulation sizes, migration rates, ancestral locations.)
- ClonalFrame/ClonalOrigin
- Infer bacterial Ancestral Recombination Graphs (generalizations of trees when recombination is present).
... there are many others!
Summary
- Bayesian phylogenetic inference is generally done by adding a tree prior to the tree likelihood. This implicitly assumes neutral evolution.
- Tree priors link the shape of the tree to parameters such as speciation rates, extinction rates, sampling rates, population sizes and migration rates.
- Development of these priors is a particularly rich area of current research.
- Due to the immense size of the state spaces involved and the use of distributions which are analytically intractable, almost all Bayesian phylogenetic inference uses MCMC.
- Deciding on stopping criteria is difficult. Many necessary requirements for convergence, but no sufficient requirements!
- Methods for summarizing sampled tree distributions exist, but may be misleading. At the moment, sticking to summary statistics for particular characteristics of interest is the only safe option.
Recommended Reading
- Chapter 10: Bayesian phylogenetic inference
Also recommended:
- Felsenstein (2003), Inferring Phylogenies
- Drummond & Bouckaert (2015), Bayesian Evolutionary Analysis with BEAST